3.1 Phenotypic characterization and PGP activity
Strains designated HM01, HM02, and HM04 were identified as M. morganii based on MALDI-TOF mass spectrometry analysis. These strains were isolated from the rhizosphere soils of chickpea, maize, and wheat fields, respectively. The isolates were further tested in detail for PGP characteristics through in vitro screening such as phosphate, potassium, and zinc solubilization, IAA production, and nitrogen fixation. All three Morganella isolates showed positive outcomes for PGP characteristics. Phosphate solubilization indices for HM01, HM02, and HM04 were found to be 4.5, 2.6, and 3.9. All the strains were able to solubilize potassium chloride as a sole source of potassium. The potassium solubilization indices were found to be 6.5 for HM01, 5.5 for HM02, and 4.2 for HM04. Zinc solubilization indices were reported to be 2.75, 4.5, and 5.1, respectively. IAA plays a major role in plant growth promotion and development, and it was observed that all the rhizosphere-associated M. morganii strains produced high levels of auxin when supplemented with L-tryptophan. The IAA concentrations were found to be 9.85, 10.36, and 9.29 µg/mL for HM01, HM02, and HM04, respectively. Nitrogen fixing ability was also confirmed from increased OD600nm in Burk's medium, in which all three isolates proved to be positive. The OD was found to be increased from 0.1 OD/mL to 1.1, 0.8 and 0.6 OD/mL for HM01, HM02, and HM04, respectively. Additionally, the strain HM02 and HM04 exhibited antifungal activity. This suggest that these strains may produce bacteriocin (compounds with antimicrobial activity), that protects the plant by inhibiting the growth of phytopathogenic fungi as well as reduces the disease-related damage (Nazari and Smith 2020). Overall PGP potential of the isolates is provided in Table 1.
Table 1
PGP activities of Morganella strains isolated from rhizosphere soil of chickpea, maize, and wheat fields.
| Sr. No. | PGP trait | HM01 | HM02 | HM04 |
| 1 | P-Solubilisation index | 4.5 | 2.6 | 3.9 |
| 2 | K-Solubilisation index | 6.5 | 5.5 | 4.2 |
| 3 | Zn-Solubilisation index | 2.75 | 4.5 | 5.1 |
| 4 | IAA production (µg/mL) | 9.85 | 10.36 | 9.29 |
| 5 | Nitrogen Fixation (OD) | 1.1 | 0.8 | 0.6 |
| 6 | Antifungal activity | - | + | + |
3.2 Genomic characterization
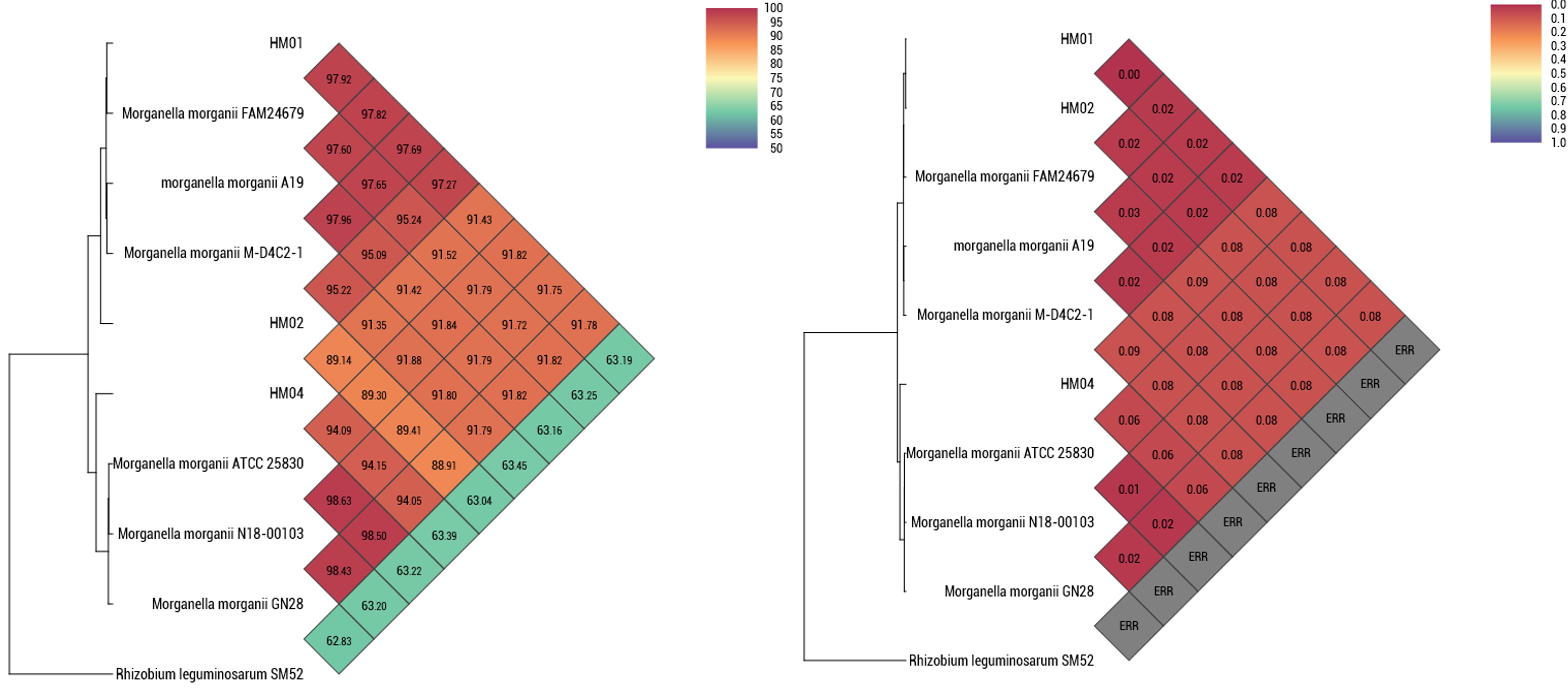
The presence and distribution of single-copy marker genes in the genomes gives ideas about genome quality. Completeness and contamination levels of rhizosphere soil-isolated Morganella genomes were calculated based on the presence, absence, or possible duplication of these conserved marker genes. It indicates possible contamination added during the DNA extraction or sequencing step. The estimated genome completeness scores were found to be 100% for HM01, 99.37% for HM02, and 92.91% for HM04, which indicate the high-quality assembly for downstream analysis. 16S rRNA-based and whole-genome-based phylogenetic trees confirmed the close genetic relationship of HM01, HM02, and HM04 to Morganella morganii ATCC 25830. Furthermore, pairwise genomic similarity analyses using ANI and dDDH methods reinforced these findings. Particularly, ANI (based on OrthoANI), and GGDC estimates all indicated the greatest similarity among the three isolates and reference strains M. morganii ATCC 25830, thus validating their species-level identification and genetic similarity (Online Resource ESM_1).
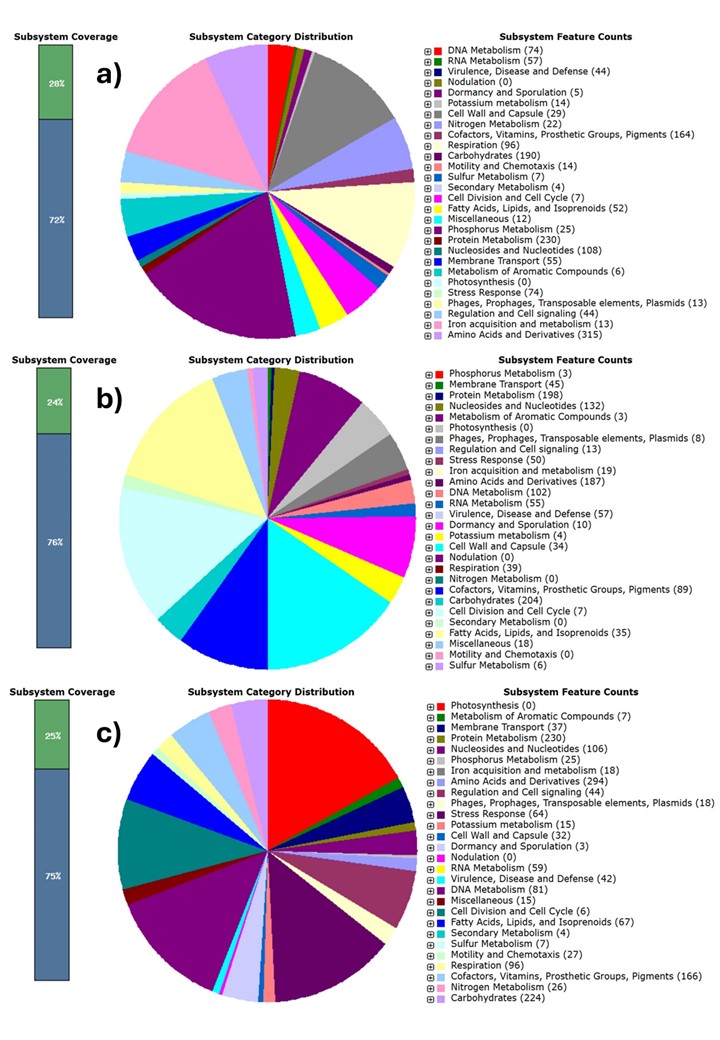
The basic genomic characteristics of rhizosphere soil-isolated Morganella strains are depicted in Table 2. Briefly, strain HM01 represents a complete circular chromosome with the highest assembly quality and an L50 value of 1. HM04 also showed a high-quality assembly, with only 13 contigs and an N50 value of 2,599,043 bp. In contrast, strain HM02 showed 330 contigs and a lower N50 value of 21,387 bp. The GC content of strains HM01, HM02, and HM04 was found to be 50.3, 41.7, and 50.6, respectively. The genome sizes of the three strains also varied from 3.8 to 4.3 Mb, with HM04 being the largest one. Corresponding with its size, HM04 showed a high count for predicted coding sequences (4,738) compared to HM01 (4,239) and HM02 (4,214). The RNA gene count was found to be 103, 94, and 103 in HM01, HM02, and HM04, respectively. Functional subsystem classification revealed a broader metabolic and functional repertoire in HM01 (333 subsystems) and HM04 (334), as opposed to HM02 (253), which could be due to its lower genome completeness or smaller genome size (Online Resource ESM_2). In all the strain, a high abundance of genes involved in pathways known to promote plant growth was observed (Table 2). Specifically, strain HM01 and HM04 demonstrated significant enrichment in genes associated with phosphorus metabolism, potassium metabolism, iron acquisition and metabolism, motility and chemotaxis, and stress response. While in strain HM02, genes related to iron acquisition and metabolism and stress response were present in higher numbers. Collectively, these observations showed that rhizosphere-associated strains possess PGP traits for both direct mechanisms (such as nutrient solubilization and uptake) and indirect mechanisms (such as stress mitigation and competitive colonization).
Table 2
Basic genomic details of rhizosphere soil-isolated Morganella strains.
| Features | HM01 | HM02 | HM04 |
| Taxonomy | Bacteria; Pseudomonadati; Pseudomonadota; Gammaproteobacteria; Enterobacterales; Morganellaceae; Morganella; Morganella morganii |
| Total data (Mb) | 4.1 | 3.8 | 4.3 |
| GC content (%) | 50.3 | 41.7 | 50.6 |
| Number of Contigs (with PEGs) | 1 | 330 | 13 |
| N50 (bp) | 4155665 | 21387 | 2599043 |
| L50 (bp) | 1 | 30 | 1 |
| Number of Coding Sequences | 4239 | 4214 | 4738 |
| Number of RNAs | 103 | 94 | 103 |
| Number of Subsystems | 333 | 253 | 334 |
| Subsystem Classification (Feature counts) |
| phosphorus metabolism | 25 | 3 | 25 |
| potassium metabolism | 14 | 4 | 15 |
| sulphur metabolism | 7 | 6 | 7 |
| iron acquisition and metabolism | 13 | 19 | 18 |
| secondary metabolism | 4 | 0 | 4 |
| motility and chemotaxis | 14 | 0 | 27 |
| Stress response | 74 | 50 | 64 |
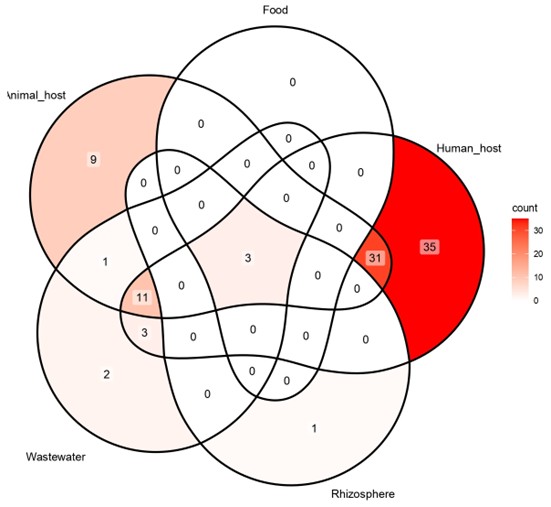
For comparative genomic analysis, a total of 84 M. morganii genomes were extracted from NCBI along with the metadata annotation on basic genomic features. Based on the metadata information, genomes lacking source information (n = 4) and those with a low CheckM completeness score and a high number of contigs (n = 2) were excluded from the study. The resulting 78 genomes were included in the study and were derived from the diverse niche, including food (n = 11), wastewater (n = 3), human clinical samples (n = 48), and various animal hosts (n = 16). Across the niche, the sizes of the genomes varied from 3.8 to 4.3 Mb. The difference in genome size was found to be mainly due to the gain and loss of functional genes (Moulana et al. 2020). The greater sizes (4.0 to 4.3 Mb) of the food- and wastewater-derived genomes suggest the acquisition of accessory genes involved in nutrient uptake, stress tolerance, and environmental adaptation. In contrast, the smaller genome sizes (3.7 to 4.0 Mb) in human- and animal-derived genomes denote streamlined genomes harbouring specialized gene sets required for the adaptation to host-associated lifestyles. Relatively stable GC content, ranging between 50.5% and 51.5% among all isolates, indicates a conserved base composition across different niches. An overview of the genomic features of the NCBI-extracted Morganella genomes is provided in Supplementary File 1.
3.3 Pan-core analysis
To characterize the genetic diversity of the Morganella, a pan-genome of soil-isolated Morganella genomes (n = 3) was performed along with publicly available genomes (n = 78), and the distribution of genomic features was analysed. Pangenome analysis revealed the distinct genomic distance between the selected Morganella strains, with the core gene alignment showing three major phylogenetic groups (Fig. 1a). The first group was the largest, well-defined cluster, consisting of 49 genomes, derived mostly from clinical and wastewater sources. The second group, consisting of soil-isolated strains HM01 and HM02, clustered separately with the food-derived isolates. Such phylogenetic allocation suggests that HM01 and HM02 could either have functional or evolutionary characteristics comparable to food-borne strains or could have analogous convergent adaptation or gene gain applicable to the food environment. This clade had a distinct block of accessory gene sets, well defined from the remaining two groups within the phylogenetic tree. Strain HM04, on the other hand, is grouped differently, closely related to the strains derived from both human as well as animal origins. The reason for this unexceptional placement is unclear; however, one possible explanation could be historical cross-environmental transmission or HGT events involving mobile genetic elements shared between host-associated environments.
Based on homology, the total gene content of the genomes was classified into three categories: core (shared by all genomes), shell (present in some genomes), and cloud/unique (found in only one genome). Pangenome analysis calculated 23829 gene clusters, with only 8.52% (2031 genes) and 13.13% (3130 genes) being classified as core- and shell-genes, respectively (Fig. 1b). A higher number of cloud genes, representing 78.34% (18668 genes) of the total pangenome, indicates that high degree of genomic flexibility in Morganella is driven by acquiring the niche-specific traits (Zhong et al. 2021). Similarly, in the study of pangenome analysis of 59 M. morganii strains, Rahman et al. (2020) observed a similarly low percentage of core genes (6.83%) and a large number of cloud genes. The pan genome curve, which plots the number of genes against the number of genomes analysed, did not reach a plateau (Fig. 1c), again indicates the open-genome nature of M. morganii. Among the studied strains, HM01, HM02, and HM04 had a combined total of 4152 unique genes, substantially more than the remaining 78 strains. Despite having a smaller genome size, strain HM02 exhibited a significant number of cloud genes (2885), followed by HM04 (1081) and HM01 (186). The remarkably high percentage of cloud genes in these strains may reflect the HGT events that act as an evolutionary force and facilitate the selection of novel functions and increase the adaptive potential of bacteria (Woods et al. 2020). Investigation of bacterial traits involved in rhizosphere colonization revealed that HGT events play an important role in genome plasticity for rhizosphere adaptation (Lopes et al. 2016). Similarly, another study reported that bacterial strains that acquired genetic materials from genomic islands through HGT, displayed better symbiotic nitrogen fixation competency compared to the closely related strains that lacked these genomic elements (Cotta et al. 2025). A significant portion of these cloud genes encoded hypothetical proteins in HM01, HM02, and HM04 (134, 1204, and 770, respectively), which exist in an environment with a lack of any supporting evidence of in vivo expression. These conserved hypothetical proteins are encoded by a significant proportion of the bacterial genome that can act as biomarkers or essential signalling proteins in the adaptation process, including biotic and abiotic stress, interspecies communication, and environmental interactions (Chirgadze et al. 2022).
Besides the hypothetical proteins, among the genes of interest, the unique genes identified in strains HM01, HM02, and HM04 were found to code for PGP activities, highlighting their potential ecological roles in the rhizosphere. Strain HM02 exhibited 19 unique PGP traits that include a wide range of functional categories needed for promoting plant growth and refining soil fertility. Pertaining to nutrient solubilization, strain HM02 contains a large repertoire of genes for phosphate solubilization (pstB3_1, pstB3_2, pstC1, and multiple pst genes) which facilitate the breakdown of inorganic phosphate and make it available for plant uptake. Besides, the strain also possesses genes for potassium solubilization (kup_1, kup_2), zinc solubilization (zupT), iron acquisition and metabolism (dtxR, fur_2), siderophore production (fepC), assimilatory and dissimilatory nitrate/nitrite reduction (napA, nirC, norG, norR_4), which help in the breakdown of essential macronutrients and micronutrients, further enhancing its PGP potential. Additionally, genes involved in chemotaxis (cheB) that enable the strain to move towards favourable environmental conditions are also annotated.
Strain HM04 contains 34 distinct PGP genes, with most of the genes being chemotaxis-associated (32), indicative of its indirect plant growth modulation function through improved environmental sensing. Among the chemotaxis-related pathways, genes that code for chemotaxis signal transduction proteins (cheB, cheR, cheW, cheY, and cheZ) are detected. Genes for structural and motor flagellar components such as (fliD, fliE, fliF, fliG, fliH, fliI, fliJ, fliM, fliN, fliO, fliP, fliQ, fliR, fliS, fliT, and motA) were also detected. These genes represent the essential components of the bacterial two-component signalling system and are responsible for movement towards favourable stimuli (Colin et al. 2021). The presence of several subunits of the tsr genes associated with methyl-accepting chemotaxis proteins showed that strain HM04 can sense and react to the spectrum of chemical compounds in the rhizosphere (Colin et al. 2021). Furthermore, genes for assimilatory sulphate reduction (cysI and cysJ) were found, which suggest its possible function in sulfur assimilation and nutrient cycling as a part of plant health enhancement.
No specific genes were present in strain HM01 directly related to plant growth. Yet, indirect plant development-influencing genes were present. These include genes for peptide uptake (sapD), starch synthesis (glgA), protein synthesis and stress tolerance (metZ), and a two-component regulatory system component facilitating environmental signal perception and response (rcsD_1). The occurrence of these cloud genes in rhizosphere bacteria indicates their possible functions in nutrient solubilization and acquisition, metabolic adjustment, and plant-microbe interaction, all of which are the essential elements of PGP mechanisms (Kumar et al. 2022a).
3.4 Principal coordinate analysis (PCoA)
To further analyse the genomic relationships observed by the pangenome analysis, a PCoA was performed. The PCoA plot based on the Bray-Cutis distance matrix demonstrated a well-defined cluster (Fig. 2a). This clustering pattern observed in the PCoA plot was consistent with those observed in the pangenome analysis based on gene composition. Genomes of different clusters were then plotted separately to study the genetic differentiation between each cluster, which reflects the degree of intra-cluster divergence along the PCA axis. In cluster 1 (Fig. 2b), even though the clustering was tight, genomes from human, animal, and wastewater isolates created separate sub-clusters. Among human-derived Morganella genomes, Morganella from various clinical samples like blood, sputum, stool, wound, and urine showed a slight but distinct segregation on the PCoA axis. These minor spatial distinctions mirror localized genetic variations that may be motivated by site-specific selective pressures within the host (Chung et al. 2017). In addition, the co-occurrence of animal-derived genomes with those from humans indicates the overlapping gene pools and regular gene flow as a result of the selective pressure, e.g., exposure to antibiotics or HGT events (Jing et al. 2022).
On the contrary, the food-derived genomes in cluster 2 (Fig. 2c) formed a highly compact and distinct cluster, clearly separating them from the rhizosphere-associated strains. This is indicative of some crucial changes in these strains that may have occurred during their adaptation to specific habitat. In rhizosphere-associated strains, these changes might be the outcome of coevolution with plants (Iqbal et al. 2021). This observation further supports that a particular habitat, such as plant-associated habitat can exert a profound effect in determining the frequency and direction of HGT in the bacterial community. It also indicates a consistency between nucleotide sequence and hierarchical clustering patterns (Iqbal et al. 2021). These results are in line with the earlier research on Streptococcus spp., in which HGT was predominantly observed within strains that share the same habitat, further supporting the ides similar environmental pressure promote the exchange of genes between microbial communities that are suited to analogous ecological niches (Richards et al. 2014). This is indicative of a highly conserved core gene in rhizosphere-soil isolates suited for stress tolerance, heavy metal detoxification, and possibly plant-associated beneficial traits, including those related to nutrient solubilization or bioremediation. Like cluster 1, the genome in cluster 3 also showed spatial separation (Fig. 2d). Though rhizosphere-soil isolated strain HM04 is phylogenetically grouped among animal- and human-associated isolates, it is still genetically separate. This is an observation that reflects ecological pressure in the rhizosphere resulting in a specific genetic makeup, different from that noted among clinical samples.
3.5 BRIG-Based Genomic Alignment
While strains HM02 and HM04 appeared more distantly clustered on the PCoA plot based on the cluster-separated analysis, strain HM01 remained closely associated with food-derived, and to a lesser extent, animal- and wastewater-derived genomes. This indicates the highly shared genetic pools, although HM01 retains unique genetic elements that set it slightly apart. Genomic flexibility analysis enables the recognition of regions that might be related to the adaptive processes of bacteria to their specific environment (Gomes et al. 2024). Therefore, to identify genomic differences within specific regions, the BRIG plot was used to examine divergence patterns (Fig. 3). The chromosomal backbone of the strain HM01 was found to be highly conserved and aligned with the reference strain M. morganii ATCC 25830. Other strains also showed large regions of similarity (98%) with the reference strain, however, the regions of dissimilarity were distributed intermittently across the genomes, which was visualized as gaps. The gaps consistently observed in the genomes, i.e., the genes present in HM01 but absent in others, were classified as major gaps (labeled 1–8) and minor gaps (a–w). The coordinates of the gap regions were manually curated and annotated using the GenBank (gbk) file derived from the Prokka tool. Specifically, the major gaps mainly contained clusters of hypothetical proteins and MGEs (prophage integrases (e.g., IntA and IntS) and transposases) and lysozyme-related genes (e.g., RrrD).
Additionally, each major gap also contains a distinct set of genes. Gap 1 contained regulatory and metabolic elements such as LexA repressor and Na⁺-translocating NADH-quinone reductase subunit E. In Gap 2, there were genes for DNA damage response (UmuC and UmuD) and a mitochondrial ubiquinone biosynthesis methyltransferase, whereas Gap 3 coded for replication and DNA repair, such as replicative helicase, a putative HTH-type transcriptional regulator, and exodeoxyribonuclease VIII. This emphasizes the potential of HM01 for DNA damage repair and maintenance of metabolic processes under stress conditions (Fu et al. 2020; Stratton et al. 2022). Gap 4 has a membrane-bound lytic murein transglycosylase C and the toxin subunit YenB that is crucial to prevent the growth and colonization of phytopathogens in the rhizosphere (Panicker and Sayyed 2022). Gap 5 comprises carbohydrate metabolism-related genes, such as UDP-glucose modifying enzymes, glycogen synthase, teichoic acid transferase, and the sensor histidine kinase CpxA. CpxA is a sensor kinase primarily involved in envelope stress response by regulating the downstream genes of the CpxAR two-component system. Activation of this system triggers a cascade in response to envelope stress, such as antimicrobial exposure or environmental fluctuations (Cho et al. 2023). Stress response and regulatory elements, including the HipA toxin, FtsH metalloprotease, RNA polymerase-associated protein RapA, RecF, along with IS3 family transposases ISLad1 and IS911, were noted in Gap 6. HipA induces translation inhibition (Nashier 2025); FtsH maintains membrane protein homeostasis (Yi et al. 2022); RapA facilitates transcriptional recovery (Qayyum et al. 2021) RecF is involved in DNA damage repair (Sass et al. 2021); and IS elements enable the bacteria to acquire advantageous traits. Gap 7 was characterized by a full suite of arsenic resistance genes (ArsR2, ArsD, ATPase, efflux pump, and reductase) that are required to survive in an environment contaminated with heavy metals (Kumar et al. 2022b).
Besides, several minor gaps comprising fewer genes were annotated to contain a diverse array of functional genes that may play an important role in PGP activity. These genes included mannose-6-phosphate isomerase (gap a; carbohydrate metabolism), autoinducer-2 kinase (gap c; quorum sensing and interspecies communication), cell division protein DamX and a putative deoxyribonuclease (RhsC) (gap e; cellular growth and genome stability), threonine/homoserine exporter RhtA (gap h; amino acid transport), acetyltransferase (gap i; post-translational modification or detoxification); peptide transporter CstA (gap n; nutrient acquisition under stress), serine endoprotease DegS (gap o; stress-response), and serine acetyltransferase (gap r; sulfur metabolism). These genes are uniquely present in strain HM01 that help plant development by nutrient acquisition as well as in stress response. Additionally, genes related to AMR, virulence and MGEs are detected viz., phenazine antibiotic resistance protein EhpR (gap f; AMR resistance), actin cross-linking toxin VgrG1 (gap l; type VI secretion systems and bacterial virulence), IS3 family transposases (ISYps8, ISLad1, ISEc52) and a ribosomal RNA large subunit methyltransferase F (gap m; horizontal gene transfer and translation regulation), and multidrug resistance protein MdtB (gap w; AMR resistance). Genes with general cellular function, such as putative hydrolase YdeN and RNA pyrophosphohydrolase (gap u; nucleotide metabolism) and exonuclease DinG (gap b; DNA repair mechanism), were also detected. Collectively, the minor gaps reflect the broad range of genes involved in stress response, metabolism, AMR, and hence plant growth. Gaps d, g, j, k, p, q, t, and v were found to contain hypothetical proteins. This variation in gene content correlated with the finding that classifies Morganella as a highly recombinogenic bacterium that can frequently acquire genetic elements through HGT events (Jing et al. 2022).
3.6 Role of hypothetical proteins
As revealed by the pangenome and the BRIG analysis, the majority of cloud genes present in the rhizosphere soil-derived Morganella genome were classified as hypothetical proteins. These hypothetical proteins, without any supporting evidence of in vivo expression, may act as biomarkers or essential signalling proteins in the adaptation process, including responses to biotic and abiotic stress, interspecies communication, and environmental interactions (Rahman et al., 2022). Hence, the predictive functional characterization of hypothetical proteins was performed to elucidate their potential biological role. Of the 134, 1204, and 770 hypothetical proteins, functional predictions for 56, 563, and 335 were successfully obtained in HM01, HM02, and HM04, respectively.
Most hypothetical proteins annotated as endonucleases and reverse transcriptases that were associated with cellular functions such as DNA repair. Some proteins matched with DNA-binding motifs like lambda repressor-like, MerR-type, GntR-type, and winged helix-turn-helix (HTH) domains that are involved in putative regulatory activities. In all three strains, N-acetylmuramoyl-L-alanine amidases was predicted. These enzymes are peptidoglycan hydrolases that cleave the bond between N-acetylmuramic acid and L-alanine, and responsible for cell wall remodelling, cell separation, biofilm formation, and growth inhibition of phytopathogens (Guzmán-Moreno et al. 2022). Another major group of hypothetical proteins showed functional similarity with the Major Facilitator Superfamily (MFS), a group of integral membrane protein that takes part in several important processes of bacterial cell physiology. In PGP bacteria, MFS involved in nutrient transport, antimicrobial compound efflux, and cell-to-cell signalling, and contributes to increasing the plant resistance against pathogens via suppression of phytopathogens or modulation of plant defence (Pasqua et al. 2021). A similar functional role of MFS genes has been observed in plants. A genome-wide analysis of Populus trichocarpa identified 41 MFS genes (PtrMFSs) that were highly expressed in response to fungal infection (Fusarium oxysporum) (Diao et al. 2021). In addition, some of the hypothetical proteins were predicted to have stress response and virulence function-related domains, e.g., Salmonella virulence plasmid proteins encoded by the spv operon that play a role in systemic infection by increasing bacterial survival and replication inside host cells (Kang et al. 2024). Domains such as Colicin V synthesis proteins involved in bacteriocin production, phage tail fibre proteins for host recognition and virulence, and components of type IV secretion systems responsible for translocating effector proteins into host cells are also predicted (Nazari and Smith 2020). This reflects the strain’s ability to enhance bacterial competitiveness, host colonization, and adaptation to environmental stress. The complete results of the preliminary annotation of hypothetical proteins are provided in Supplementary File 2.
Hypothetical proteins with predicted role in plant growth and development were manually curated. In HM02, which harbours the largest group of hypothetical proteins, several were related to iron sequestration and metabolism (FeoA_2, iron siderophore/cobalamin periplasmic-binding domain profile, ferritin-like domain), heavy metal detoxification (heavy-metal-associated domain), and sulphur metabolism (sulfite exporter TauE/SafE). Additionally, some hypothetical proteins were predicted to be involved in stress response (glycine betaine/proline betaine transport system permease protein ProW, glyoxylase/bleomycin resistance protein/dioxygenase superfamily), cell wall modification and stress response (lipopolysaccharide choline phosphotransferase LicD, teichuronic acid biosynthesis protein TuaE), biofilm production (colanic acid biosynthesis UDP-glucose lipid carrier transferase), secondary metabolite production (S-adenosyl-L-methionine-dependent methyltransferases, acetyltransferase (GNAT) domain, radical SAM superfamily, alpha/beta hydrolase, PLP-dependent transferases), and antifungal properties (LysM domain). Furthermore, the major group of hypothetical proteins in HM02 was predicted to be involved in membrane transport. These proteins were found to be related to the sugar ABC transporter integral membrane protein, the phosphate transport system permease, the D-xylose-binding periplasmic protein, the maltose/maltodextrin transport system permease protein MalF, the PTS system sugar-specific permease components, the ECF transporter substrate-specific components, the C4-dicarboxylate anaerobic carrier, the N-acetylgalactosamine permease II component, and bacterial extracellular solute-binding proteins. These proteins help plants absorb nutrients that are beneficial to plants.
In HM04, proteins are predicted to be involved in lipopolysaccharide biosynthesis (glycosyltransferase family 25, nucleotide-diphospho-sugar transferases), carbohydrate biosynthesis (glycosyltransferase and glycosyltransferase GT-D fold), polysaccharide biosynthesis (polysaccharide biosynthesis protein), membrane transport (bacterial extracellular solute-binding protein), secondary metabolite production (acyltransferase family and acetyltransferase (GNAT) family), stress response (cyclopropane-fatty-acyl-phospholipid synthase), substrate hydrolysis (alpha/beta-hydrolases), bacteriocin production (S-type pyocin and colicin D domains), and antifungal properties (lysozyme-like domains and polysaccharide lyase). In HM01, proteins related to biofilm production (epsG family), secondary metabolite production (acyltransferase family and acetyltransferase (GNAT) family), polysaccharide biosynthesis (polysaccharide biosynthesis protein), metal ion homeostasis and oxidative stress resistance (cupin fold metalloproteins), and carbon storage and energy regulation (glycogen phosphorylase B) were predicted.
In a study, Guzmán-Moreno et al. (2022) identified several hypothetical proteins in Bacillus megaterium HgT21 that are involved in heavy metal degradation, phosphate solubilization, membrane transport and bacteriocin production. Similarly, Msimbira et al. (2022) reported that some hypothetical proteins in Lactobacillus helveticus (EL2006H) and Bacillus subtilis (EB2004S) were upregulated in response to changes in environmental conditions, such as pH. They concluded that these proteins may be expressed in response to specific environmental conditions and could play a major role in the adaptation process of bacteria. In a similar line, several studies identified a large repertoire of hypothetical proteins in strains associated with PGP activity (Guo et al. 2020; Balderas-Ruíz et al. 2020; Zhang et al. 2022). However, so far, it remains unclear whether the observed enhanced activity of the strains is due to the emergence of unique hypothetical protein or other biomolecules in PGP bacterium. Therefore, deeper insights into the predictive role of these significantly unique and potentially upregulated hypothetical proteins are required to better elucidate their involvement in plant growth and development.
3.7 AMR gene profiling
The presence-absence heatmap for the AMR gene (ARGs) identified across 81 Morganella strains is illustrated in Fig. 4. A total of 98 ARGs that confer resistance to 15 different antibiotic classes were identified. The classes include β-lactams, aminoglycosides, macrolides, lincosamides, streptogramins, fluoroquinolones, chloramphenicol, tetracyclines, trimethoprim, sulfonamides, rifampin, streptothricin, bleomycin, fosfomycin, and biocide resistance. The most prevalent ARGs were found to be from β-lactams class (27/98), followed by aminoglycosides (24/98) and macrolides (8/98). Human host-derived genomes that comprise the largest subset harbored numerous ARGs (83/98, 85%). Almost all the genomes in this niche exhibited multiple resistance determinants, with frequent detection of bla genes (blaCTX-M-15, blaCTX-M-65, blaTEM, blaSHV-12, blaKPC, blaKPC-2, blaNDM-5, blaIMP-27, blaVIM-1, blaOXA-10, blaDHA-5, blaDHA-27, and so on), conferring resistance to β-lactam antibiotics, such as penicillins, cephalosporins, Carbapenems, and Monobactams. ARGs within aminoglycoside resistance class (aac(3)-IIe, aac(6')-Ib, aac(6')-Ib3, aac(6')-Ib-cr, aadA5, aadA13, aph(3')-VIb, aph(3')-XV, rmtB1, rmtC), were present in approximately 95% of human-associated genomes. Presence of β -lactamases belonging to the AmpC b-lactamase (blaAmpC) family on the chromosome of Morganella leads to an intrinsic resistance to most β-lactam antibiotics as well as first and second- generation cephalosporins (Xiang et al. 2021). A recent study detected numerous carbapenemases in Morganella sp., including VIM-1, NDM-1, NDM-5, OXA-48, OXA-181, and OXA-641 (Bonnin et al. 2024). Additionally, in a study characterizing ARGs across 102 genomes of Morganella, Zhu et al. (2025) reported a total of 241 aminoglycoside phosphotransferase-related genes. Aminoglycoside along with β-lactam antibiotics have been extensively used to treat severe infection in human and animals from last 60 years. In recent years, these combination antibiotics have been used to treat tularemia, nosocomial surgical wound infections, sepsis, plague, blood-stream infections, central nervous system infection, endophthalmitis, endocarditis, brucellosis, urinary tract infections, pneumonia, chorioamnionitis and systemic infections caused by Morganella (Lebeaux et al. 2020). However, the long-term use of these antibiotics caused the acquisition and dissemination of aminoglycoside and β-lactam resistant genes in various clinical isolates, such as Escherichia, Acinetobacter, Salmonella, Klebsiella, as well as in Morganella (Wang et al. 2022).
ARGs conferring resistant to trimethoprim (dfrA1, dfrA12, dfrA27, dfrA17, dfrA14, dfrA42, dfrA19), sulfonamide (sul1, sul2, sul3), tertracycline (tet(A), tet (B), tet(D)), chromaphenicol (catA1, catA2, catB3, catB8, floR, cmlA1, cmlA5, catB2), were also detected in 85% of the clinical genomes in present study. Bonnin et al. (2024) reported that intrinsic resistance to tetracycline was observed only in M. sibonii, while the tetracycline resistance genes was found in 42 M. morganii isolates in the present study. Other ARGs identified in ~ 70% of genomes included fosfomycin (fosA3), bleomycin (ble, bleO), macrolid (mph(A), mph(E), ere (B)), fluroquinolone (qnrS1), microlid-streptomycin B efflux genes (msr(E)). Genes conferring resistance to biocide (qacEdelta1, qacL), rifampin (arr, arr-2, arr-3), lincosamide (lnu(F), lnu(G)), and streptothricin (sat2) were also detected in some of the Morganella genomes which were isolated from the patients subjected to multiple antibiotic treatments. This observation is in line with the large-scale genomic study conducted on clinical isolates of Morganella which reported a high rate of ARGs and the emergence of multidrug-resistant strains (Zhu et al. 2025). The co-occurrence of multiple ARGs suggests a multidrug-resistant phenotype, which was driven by the intense use of antibiotics in healthcare settings. These indicate that Morganella from hospitalized patients had evolved to acquire many more ARGs to encounter complex and high selection of antimicrobials in the clinical environment.
Genome clustering with respect to the ARG profiles was observed that indicates that each genome in the specific niche acquired different ARGs (Fig. 4). Among the 83 ARGs found in the human-derived genomes, 43 ARGs were shared with animal-derived ones (Online resource ESM_3). In a similar line, Jing et al. (2022) reported 34 acquired ARGs shared between Morganella isolates from both sources, indicating a long history of acquisition and widespread dissemination of these genes within the genus. The ARGs dissemination between these habitats can occur through multiple routes such as food animals, direct contact between humans and animals, or through shared environmental resources, such as contaminated water, with latter being the most common source (Cao et al. 2022). Similarly, a present study observed that Morganella genomes which were derived from the wastewater shared 14 ARGs with human-derived genomes, 11 of which were also detected in animal-derived genomes, indicating that wastewater acts as a convergence point for ARGs dissemination between different habitats (Hutinel et al. 2022). Additionally, animal-derived genome exhibited 9 unique ARGs belonging to β-lactam (blaCTX-M-63), aminoglycoside (aph(3')-VI), trimethoprim (dfrA24, dfrA23, dfrA10), chloramphenicol (cmlA6), macrolide (erm(42)), biocide (qacE), fluoroquinolone (qepA). Two unique ARGs, namely aac(6')-Ie and tet(L) were also detected in wastewater.
A total of 3 ARGs (blaDHA, catA2, and tet (D)), involved in resistance to 3 different categories of antimicrobials, were shared among Morganella isolates from all the niche, indicating a core set of resistance determinants that persist regardless of the environment (Online resource ESM_3). These were also the only ARGs detected in food and rhizosphere isolates, further suggesting that isolates from these habitats carry fewer ARGs compared to clinical or wastewater settings. Additionally, msr(C)—which confers resistance to macrolide was detected in the rhizosphere soil-derived HM02 strain. Macrolide, a critically important human medicine, enters into the soil through the application of biosolids that are applied as an organic fertilizer (Brown et al. 2022). These biosolids are frequently contaminated with pharmaceutical residues that persisted during wastewater treatment and partitioned into the organic phase. As a result, soil microbial communities may acquire resistance to macrolide to thrive in such contaminated environments. The low count of ARGs in food and rhizosphere isolates indicates lower selective pressure for resistance in these niches; however, the detection of any ARGs may raise concerns regarding food safety and environmental contamination.
3.8 Association between ARGs and mobile genetic elements
Morganella accumulated both intrinsic and acquired ARGs, leading to a multidrug resistance strain. Different types of MGEs viz., insertion elements, transposons (composite transposons, unit transposons, and Miniature Inverted-repeat Transposable Elements (MITEs)), and integrons (integron-01 and integron-02) responsible for acquired ARGs were identified in the studied Morganella genomes (Fig. 5). Insertion sequences (IS) are found to be majorly involved in the mobilization and dissemination of different ARGs. Of the 32 types of insertion elements detected; the most frequent IS families found were: IS26, IS6100, IS5057, ISAba1, ISAba125, ISCfr1, ISEc59, ISSen9, ISVsa3, ISVsa5. Among them, IS26 found to be a predominant insertion sequence, primarily associated with ARGs conferring resistance to β-lactams and aminoglycosides. ISVsa5 was found to be strongly associated with the mobilization of tetracycline resistance genes, while ISVsa3 played a crucial role in facilitating HGT events contributing to resistance against sulfonamides and fluoroquinolones. Additionally, ISAba1, ISAba125, ISCfr1, ISEc59, and ISSen9 were frequently detected in the flanking regions of ARGs conferring resistance to fluoroquinolones, biocides, aminoglycosides, sulfonamides, and chloramphenicol, respectively. This widespread association of different IS families with various classes of ARGs indicates the important role of insertion elements in facilitating the horizontal dissemination and diversification of ARGs.
Among integrons, integron-01 (characterized by the presence of the intl1 gene) represents a strong linkage with the ARGs flanking region. Major repertoire of genes linked to aminoglycoside resistance, were found to be horizontally disseminated by integron-01. Additionally, other resistance determinants such as those conferring resistance to sulphonamide, chromaphenicol, remapping, and biocide were also commonly associated with integron-01. Furthermore, trimethoprim resistance genes and quinolone resistance genes also displayed linkages with integrons, although at a lower frequency compared to aminoglycoside- and sulfonamide-resistance genes. Resistance genes against macrolides, phenicols, and rifampicin were also found to be associated with integrons-01, although these connections were less numerous. Furthermore, qacEdelta1 that is known as a multidrug efflux gene were flanked by the class 1 integron, further contributing to enhanced resistance ability. However, the role of integron-02 (characterized by the presence of the intl2 and intl3 genes) in mobilization of ARGs was limited and found to be associated with only few genes, such as catB3, qacEdelta1, and arr.
Transposons were also found to be involved in the dissemination of various ARGs, although their associations were less frequent compared to insertion elements and integrons. Among the three types of transposons identified, composite transponsons are found to be more associated with the dissemination of ARGs. Composite transposons associated with IS26 were most frequent and associated with resistance genes against β-lactams, aminoglycosides, sulfonamide, bleomycin, and fosfomycin. Unit transposons, on the other hand, were mainly associated with resistance to macrolides and chloramphenicol, while MITEs were occasionally linked to β-lactam, aminoglycoside, and bleomycin resistance to a much lesser extent. It should be noteworthy that these MGEs are found in the flanking region of the ARGs which was predominant in the Morganella genomes isolated from human, animal, and wastewater. Interestingly, ARGs detected in the food and rhizosphere soil isolates were not found to be associated with any MGE.
In the investigation of ARGs linked MGEs, a large repertoire of MGEs were detected in studied Morganella genomes. This analysis also revealed that more than one type of MGEs are involved in dissemination of resistance against particular ARG. In a study investigating the evolutionary trends of Morganella, Chen et al. (2024) reported that M. morganii undergoes evolution driven by MGEs, which significantly enhance its adaptability to environmental changes and the selective pressures imposed by clinical antimicrobial agents. Additionally, Xiang & Li, (2021) characterize two novel mobile genetic elements (Tn6835 and MMGI-1) in a Morganella strain isolated from fecal swab of healthy chicken and found that most of the ARGs were located on these MGEs which are responsible for pan-resistant nature of Morganella against all known antibiotics. Additionally, few studies have revealed new transposons like Tn7376 in Morganella and genomic islands that have multidrug resistance genes, like dfrA24, enabled by IS26-mediated recombination (Jing et al. 2022; Luo et al. 2022). Dissemination of blaKPC-2 and blaNDM-1 in Carbapenem-resistant M. morganii (CRMM) isolates was also found to be majorly facilitated by IncL/M plasmids and IS26-mediated transposon activity (Yao et al. 2025).
In the circos plot, the color coding on the ring denotes the location of ARGs. Most of the ARGs associated with MGEs are found to be plasmid-borne. For those with are located on both plasmid and chromosome, their occurrence was less frequent on the chromosome. Plasmid-derived ARGs were identified from species such as Escherichia coli, Proteus mirabilis, Citrobacter freundii, Aeromonas rivipollensis, Pseudomonas aeruginosa, Salmonella enterica, Pasteurella aerogenes, Enterobacter cloacae, and Acinetobacter baumannii. AMR pangenome profiling of the 827 genomes from Enterobacteriaceae family, collected from livestock farms and wastewater, identified distinct dynamics for chromosomal and plasmid-borne ARGs. They found that plasmids carry a substantial burden of AMR genes and MGEs. Furthermore, AMR-gene-carrying plasmids appear to be under stronger selective pressure and are primarily responsible for conferring resistance to multiple antibiotics, specifically in clinical strain compared to non-clinical ones. These findings indicate that clinical isolates act as major reservoirs for the transfer of ARGs into Morganella species. Although direct evidence of ARG transmission from clinical isolates to M. morganii via mobile genetic elements is limited, several studies have reported the presence of clinically relevant resistance genes on plasmids and transposons in M. morganii isolates, suggesting potential acquisition through the clinical mobilome (Yao et al. 2025). Additionally, in a study by Sugita et al. (2022), it was found that inter-plasmid transposition of Tn4401a facilitates horizontal transfer of blaKPC-2 from Klebsiella pneumoniae to M. morganii through ColRNAI plasmids, enhancing resistance spread. On the other hand, ARGs that were located on the chromosome, generally not associated with MGEs and were primarily found in isolates originating from food and rhizosphere environments. This suggests that Morganella when present in non-clinical environment, such as food or rhizosphere, where the AMR pressure is low, lacks these ARGs and instead showed beneficial traits responsible for plant growth or environmental survival.
3.9 PGP pathway analysis
The genome sequences of the rhizosphere-soil isolated strains were evaluated for their ability to improve plant development through both direct and indirect effects. The direct effects involve the pathways for nitrogen fixation, phytohormone production, and solubilization of nutrients such as phosphate, potassium, zinc, sulphate, and iron. In contrast, the indirect pathway involves the suppression of pathogenic growth and colonization, degradation of aromatic and toxic compounds, quorum sensing, biofilm formation, and the production of bacteriocins and secondary metabolites. Concerning the direct pathway, a total of 33, 12, and 38 genes related to nutrient solubilization were detected in strains HM01, HM02, and HM04, respectively (Table 3). The presence of phosphate, potassium, and zinc solubilization machinery in the strain correlated with the in-vitro activities. After the application of fertilizers in soil, a major portion of inorganic nutrients remains immobilized, leaving them unavailable for plants. Therefore, it is important for the soil microbial community to produce enzymes and organic acids to solubilize this poorly soluble mineral nutrients. Besides, these compounds must be transported across the plasma membrane before they may be used (Kumar et al. 2022a). In rhizosphere-associated strain genes related to phosphate solubilization such as phnV (hydrolyse phosphonate into phosphate and alkane) and pstBACS (phosphate transporter) were detected. Moreover, genes involved in the potassium (kdp, kup), zinc (znt, znu, zup, zur) and sulphate (cys) transport and uptake were also annotated in these strains. High-affinity iron chelating compounds produced by the microbial community helps in collecting iron from the soil (Kumar et al. 2022a). All strains were able to synthesis enterobactin sideophore (fep, fpt, ent) which are responsible for recovery of siderophore from the complex environment, while the protein (fhu) that help in transport and binding of iron are only detected in HM01 and HM04. Moreover, genes related to iron sequestration and metabolism were detected in all three strains (feo, efe, fur, fec, fpt, dtx).
Chemotaxis genes, which play a major role in stress response, were most abundant in HM04 (30), followed by HM01 (17) and HM02 (11). These genes include clusters such as fli (fliDEFGHIJMNOPQRST), mot (motAB), che (cheABRWYZ), as well as aer, dppA, rbsB, tap, tar, tsr, and trg, all of which contribute to endophytic traits such as chemotactic movement and host attachment. Nitrogen metabolism represents another important pathway that promotes plant growth and biomass accumulation. Genes related to the nitrogen fixation pathway were not detected in any of the isolates. However, genes involved in the indirect nitrogen metabolism pathway, such as nitrification-denitrification (narG, narH, narI, norG, norR, napA) and assimilatory/dissimilatory nitrate reduction (nasD, narG, narH, narI, narJ, narK, napA), as well as nitrite reduction (nirC), were annotated. The overall presence-absence gene matrix for these three isolates involved in both direct and indirect PGP activity is shown in Table 3.
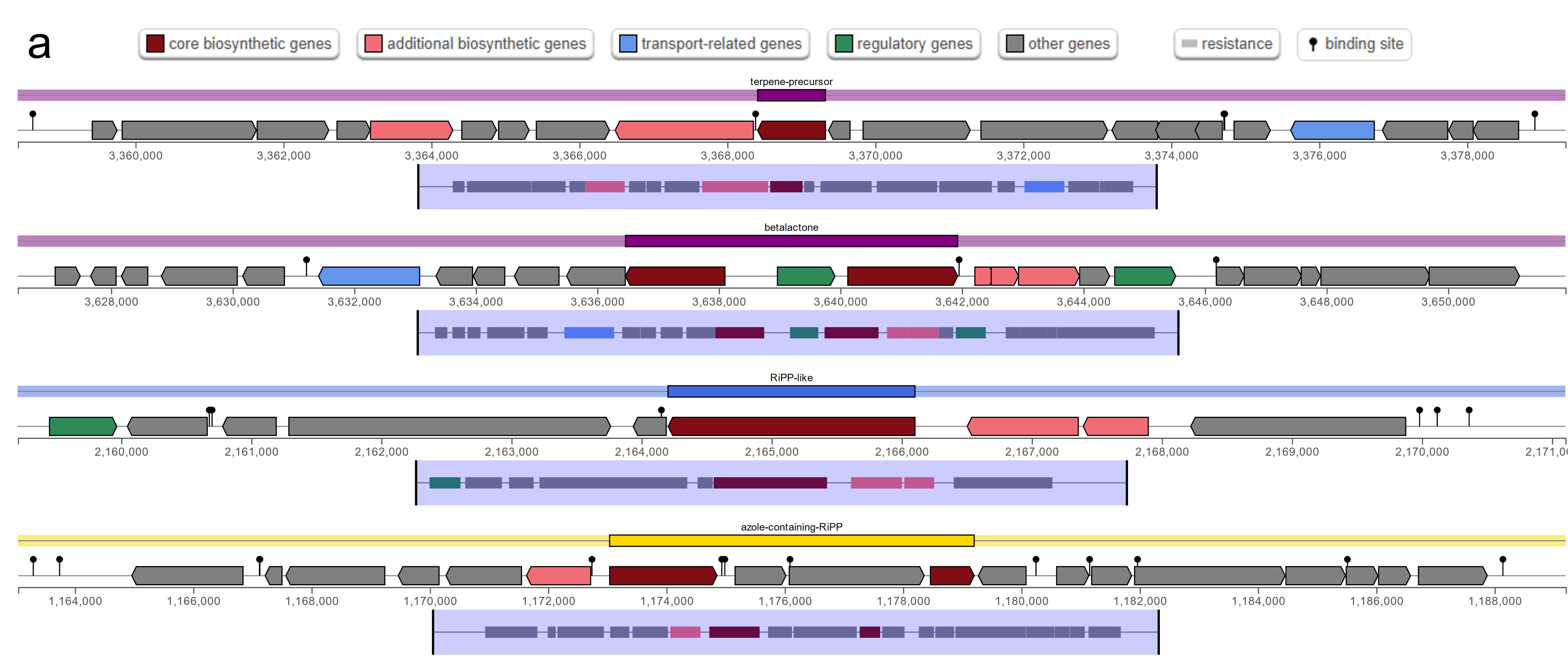
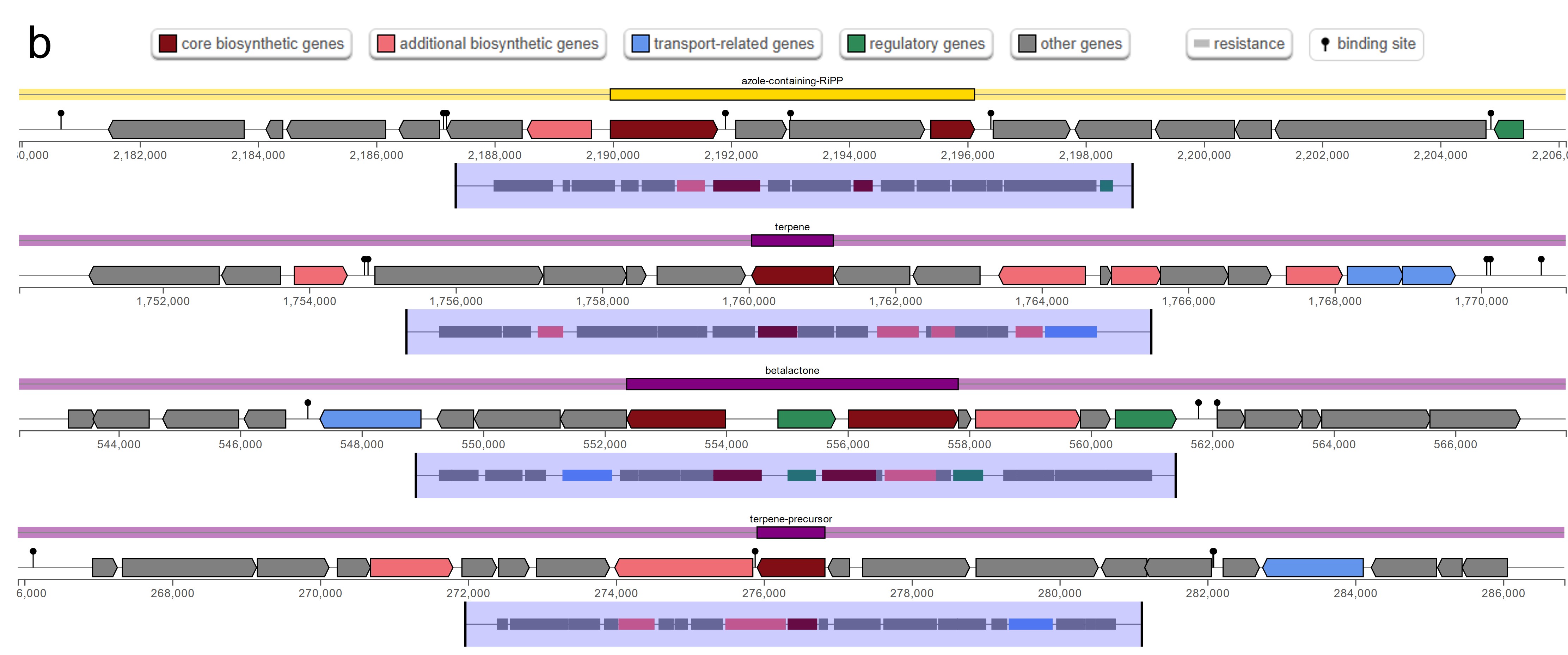
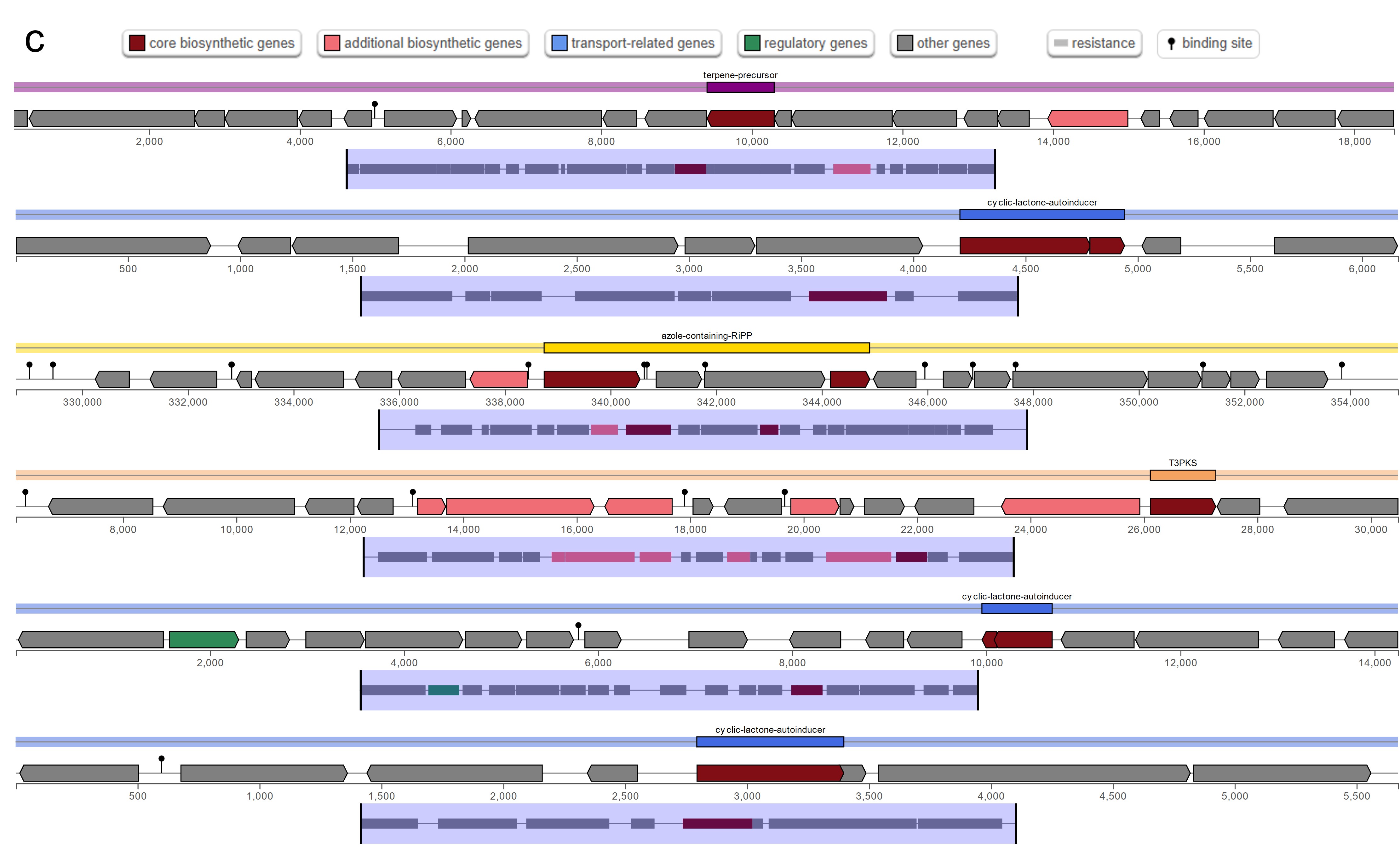
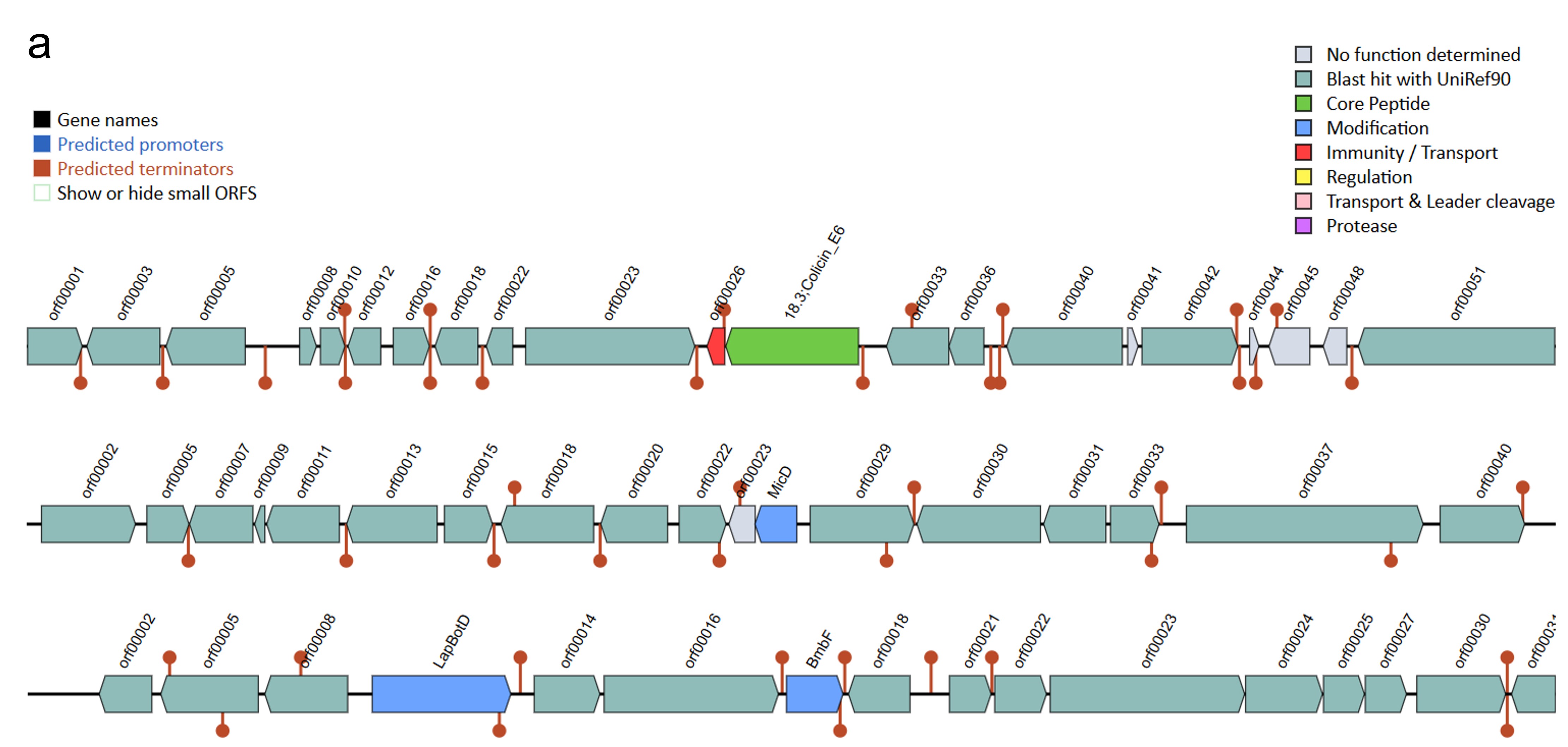
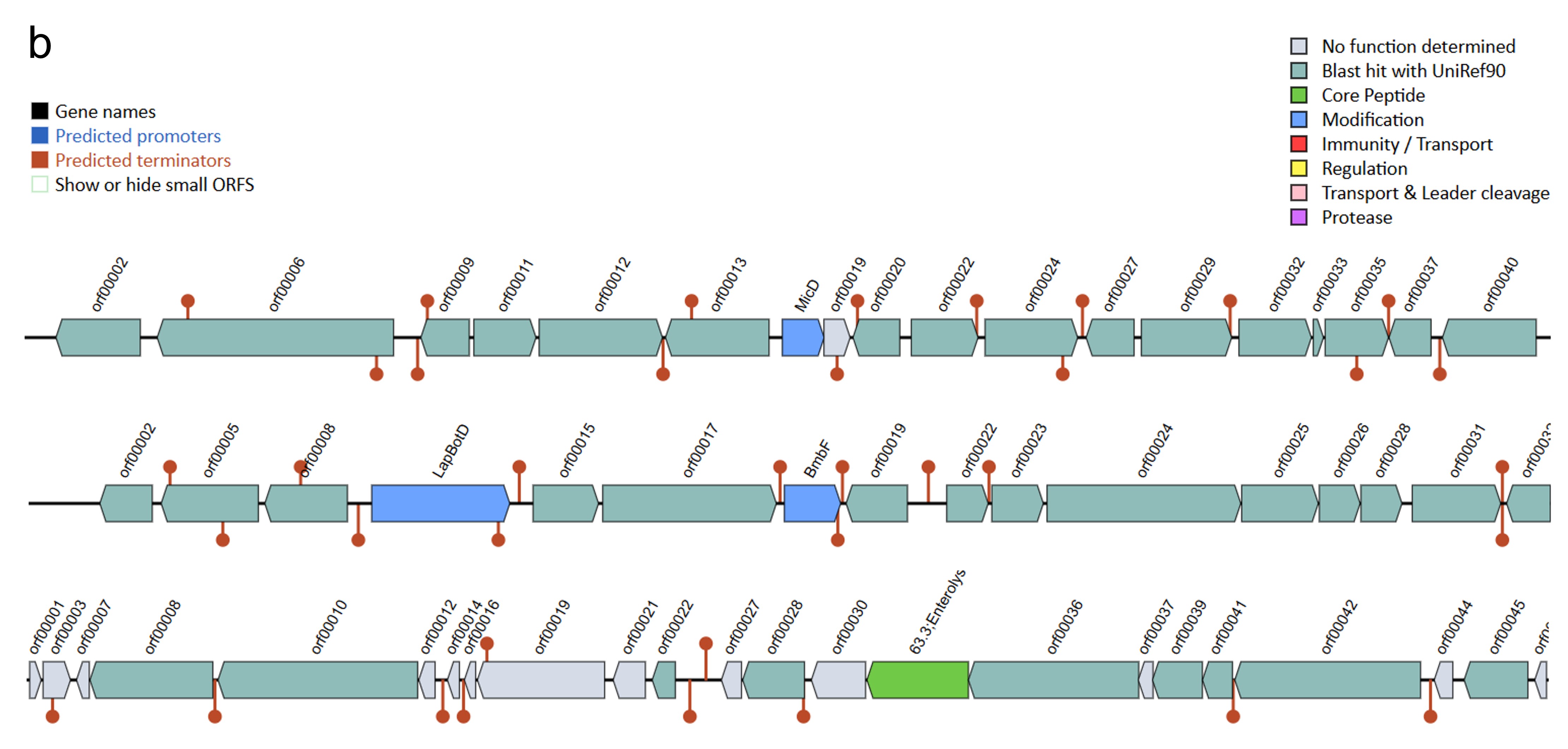
The antiSMASH analysis revealed a total of five, six, and five secondary metabolite biosynthetic gene clusters in strains HM01, HM02, and HM04, respectively (Online resource ESM_4). Two clusters, namely the azole-containing RiPP (antimicrobial activities) and terpene precursor clusters (production of bioactive compounds for signaling and defense), were found in all three strains. The azole-containing RiPP was previously reported to produce diverse antimicrobial peptides that inhibit the growth of phytopathogenic fungi and bacteria (Thamvithayakorn et al. 2025). Terpene is generally considered to be a fungal and plant natural product. However, a study identified that bacterial genomes also harbor gene cluster encoding for terpenes and produce bioactive compounds for signaling and defense (Yamada et al. 2015). The β-lactone cluster that are known for antimicrobial and anticancer properties (Awolope et al. 2021) was detected in HM01 and HM04, while cyclic lactone autoinducer that induces quorum sensing and regulation of microbial community behaviours (Kapadia et al. 2022) was uniquely observed in HM02. Furthermore, four types of bacteriocins were annotated (Online resource ESM_5). Microcin and Bottromycin were found in all three strains, whereas Colicin_E6 and Enterolysin_A were uniquely found in HM01 and HM02, respectively. These bacteriocins are potentially responsible for antagonizing the growth of phytopathogens, as reported previously (Nazari and Smith 2020).
Table 3
Functional annotation of genes associated with PGP pathways in strain HM01, HM02, and HM04.
| Pathway | Gene | Function | HM01 | HM02 | HM04 |
| Phosphate solubilization | phnV | transport system permease protein responsible for 2-ami0ethylphosphonate | ● | ○ | ● |
| pstA | Phosphate transport system permease protein pstA | ● | ○ | ● |
| pstB | Phosphate-import ATP-binding protein pstB | ● | ● | ● |
| pstC | Phosphate transport system permease protein pstC | ● | ● | ● |
| pstS | Phosphate-binding protein pstS | ● | ○ | ● |
| potassium solubilization | kdpA | High-affinity K + transport protein A | ● | ○ | ● |
| kdpB | High-affinity K + transport protein B (ATPase) | ○ | ○ | ● |
| kdpC | High-affinity K + transport protein C | ○ | ○ | ● |
| kdpD | Sensor histidine kinase (KdpD) | ○ | ○ | ● |
| kup | Low-affinity K + transporter (Kup system) | ○ | ● | ○ |
| Zinc solubilization | zntA | Zinc/cadmium/lead-transporting P-type ATPase | ○ | ○ | ● |
| zntB | Zinc transport protein ZntB | ● | ○ | ● |
| zntR | HTH-type transcriptional regulator ZntR | ● | ○ | ● |
| znuA | High-affinity zinc uptake system protein ZnuA | ● | ○ | ● |
| znuB | High-affinity zinc uptake system membrane protein ZnuB | ● | ○ | ● |
| znuC | Zinc import ATP-binding protein ZnuC | ● | ○ | ● |
| zur | Zinc uptake regulation protein | ● | ○ | ● |
| zupT | low-affinity zinc-uptake system | ○ | ● | ○ |
| Iron sequetration and metabolism | efeO | Iron uptake system component EfeO | ○ | ○ | ● |
| feoA | Fe(2+) transport protein A | ● | ○ | ● |
| feoB | Fe(2+) transporter FeoB | ● | ○ | ● |
| feoC | putative [Fe-S]-dependent transcriptional repressor | ● | ○ | ● |
| fur | Ferric uptake regulation protein | ● | ● | ● |
| yggX | putative Fe(2+)-trafficking protein | ● | ● | ● |
| fecA | Fe(3+) dicitrate transport protein FecA | ● | ○ | ○ |
| fptA | Fe(3+)-pyochelin receptor | ● | ○ | ○ |
| dtxR | Ferric uptake regulation protein | ○ | ● | ○ |
| siderophore production | fepA | Ferrienterobactin receptor | ● | ○ | ● |
| fhuC | Iron (3+)-hydroxamate import ATP-binding protein | ● | ○ | ● |
| fptA | Ferrienterobactin receptor | ● | ○ | ○ |
| fhuB | Iron (3+)-hydroxamate import system permease protein | ○ | ○ | ● |
| entS | Enterobactin exporter | ○ | ○ | ● |
| fepC | Ferric enterobactin transport protein FepC | ○ | ● | ○ |
| Assimilatory sulphate reduction | cysA | Sulfate/thiosulfate import ATP-binding protein CysA | ● | ● | ● |
| cysE | Serine acetyltransferase | ● | ○ | ● |
| cysG | Siroheme synthase | ● | ○ | ● |
| cysI | Sulfite reductase [NADPH] hemoprotein beta-component | ● | ○ | ● |
| cysL | HTH-type transcriptional regulator CysL | ● | ○ | ● |
| cysP | Thiosulfate-binding protein | ● | ● | ● |
| cysQ | 3? (2? ),5? -bisphosphate nucleotidase CysQ | ● | ○ | ● |
| cysS | CysteineÐtRNA ligase | ● | ○ | ● |
| cysT | Sulfate transport system permease protein CysT | ● | ● | ● |
| cysW | Sulfate transport system permease protein CysW | ● | ● | ● |
| cysZ | gulator CysL cysZ S | ● | ○ | ● |
| cysJ | Sulfite reductase [NADPH] flavoprotein alpha-component | ○ | ○ | ● |
| chemotaxis | aer | aerotaxis receptor | ● | ○ | ● |
| cheA | two-component system, chemotaxis family, sensor kinase CheA | ● | ● | ● |
| cheB | two-component system, chemotaxis family, protein-glutamate methylesterase/glutaminase | ● | ● | ● |
| cheR | chemotaxis protein methyltransferase CheR | ● | ● | ● |
| cheW | purine-binding chemotaxis protein CheW | ● | ● | ● |
| cheY | two-component system, chemotaxis family, chemotaxis protein CheY | ● | ● | ● |
| cheZ | chemotaxis protein CheZ | ● | ● | ● |
| dppA | dipeptide transport system substrate-binding protein | ● | ○ | ● |
| fliG | flagellar motor switch protein FliG | ● | ○ | ● |
| fliM | flagellar motor switch protein FliM | ● | ○ | ● |
| fliN | flagellar motor switch protein FliN | ● | ○ | ● |
| motA | chemotaxis protein MotA | ● | ● | ● |
| motB | chemotaxis protein MotB | ● | ● | ● |
| rbsB | ribose transport system substrate-binding protein | ● | ○ | ● |
| tap | methyl-accepting chemotaxis protein IV, peptide sensor receptor | ● | ○ | ● |
| tar | methyl-accepting chemotaxis protein II, aspartate sensor receptor | ● | ● | ● |
| tsr | methyl-accepting chemotaxis protein I, serine sensor receptor | ● | ● | ● |
| trg | methyl-accepting chemotaxis protein III, ribose and galactose sensor receptor | ○ | ● | ● |
| fliD | flagellar motor switch protein FliD | ○ | ○ | ● |
| fliE | flagellar motor switch protein FliE | ○ | ○ | ● |
| fliF | flagellar motor switch protein FliF | ○ | ○ | ● |
| fliH | flagellar motor switch protein FliH | ○ | ○ | ● |
| fliT | flagellar motor switch protein FliT | ○ | ○ | ● |
| fliJ | flagellar motor switch protein FliJ | ○ | ○ | ● |
| fliO | flagellar motor switch protein FliO | ○ | ○ | ● |
| fliI | flagellar motor switch protein FliI | ○ | ○ | ● |
| fliP | flagellar motor switch protein FliP | ○ | ○ | ● |
| fliQ | flagellar motor switch protein FliQ | ○ | ○ | ● |
| fliR | flagellar motor switch protein FliR | ○ | ○ | ● |
| fliS | flagellar motor switch protein FliS | ○ | ○ | ● |
| Nitrification | narG | nitrate reductase / nitrite oxidoreductase, alpha subunit | ● | ○ | ● |
| narH | nitrate reductase / nitrite oxidoreductase, beta subunit | ○ | ○ | ● |
| Denitrification | narG | nitrate reductase / nitrite oxidoreductase, alpha subunit | ● | ● | ● |
| narH | nitrate reductase / nitrite oxidoreductase, beta subunit | ○ | ● | ● |
| narI | nitrate reductase gamma subunit | ● | ● | ● |
| 0rG | Regulator of nitric oxide reductase genes | ○ | ● | ○ |
| 0rR | Transcriptional activator responding to nitric oxide | ○ | ● | ○ |
| | napA | nitrate reductase (cytochrome) | ○ | ● | ○ |
| Assimilatory nitrate reduction | nasD | nitrite reductase [NAD(P)H] large subunit | ● | ● | ● |
| Dissimilatory nitrate reduction | narG | Respiratory nitrate reductase 1 alpha chain | ● | ○ | ● |
| narH | Respiratory nitrate reductase 1 beta chain | ○ | ○ | ● |
| narI | Respiratory nitrate reductase 1 gamma chain | ● | ○ | ● |
| narJ | Nitrate reductase molybdenum cofactor assembly chaperone narJ | ● | ○ | ● |
| narK | Nitrate/nitrite transporter nark | ● | ○ | ● |
| napA | nitrate reductase (cytochrome) | ○ | ● | ○ |
| Dissimilatory Nitrite Reduction | nirC | Nitrite transporter | ○ | ● | ○ |
● represent gene presence
○ represent gene absence
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}