2.1 Ethics Considerations
Ethical approval was obtained from the Osun State University Ethical Review Committee, College of Health Sciences (UNIOSUNHREC 2025 /002B).
Every effort was made to minimize discomfort to animals throughout the study. The animals were maintained in sanitized, well-ventilated cages with unrestricted access to food and safe water ad libitum. All sacrifices were performed via physical methods on the anaesthetized animals, to ensure that chemical agents would not interfere with results. Hence, cervical dislocation involving a rapid, forceful separation of the first cervical vertebra from the skull was considered (American Veterinary Medical Association). This method was chosen to minimize distress and is consistent with internationally accepted protocols for small rodents.
Informed consent was obtained from poultry vendors before sample purchase. Sample collection followed national biosafety and ethical guidelines; no additional permits were required for market-purchased poultry products as all samples were sourced from legally registered markets, and collection complied with national guidelines for handling food-origin materials. No endangered or protected species were involved.
2.2 Study Location
This study was conducted at the Department of Microbiology Laboratory and Animal house, Osun State University, Osogbo, Nigeria.
2.3 Sample Collection and Processing
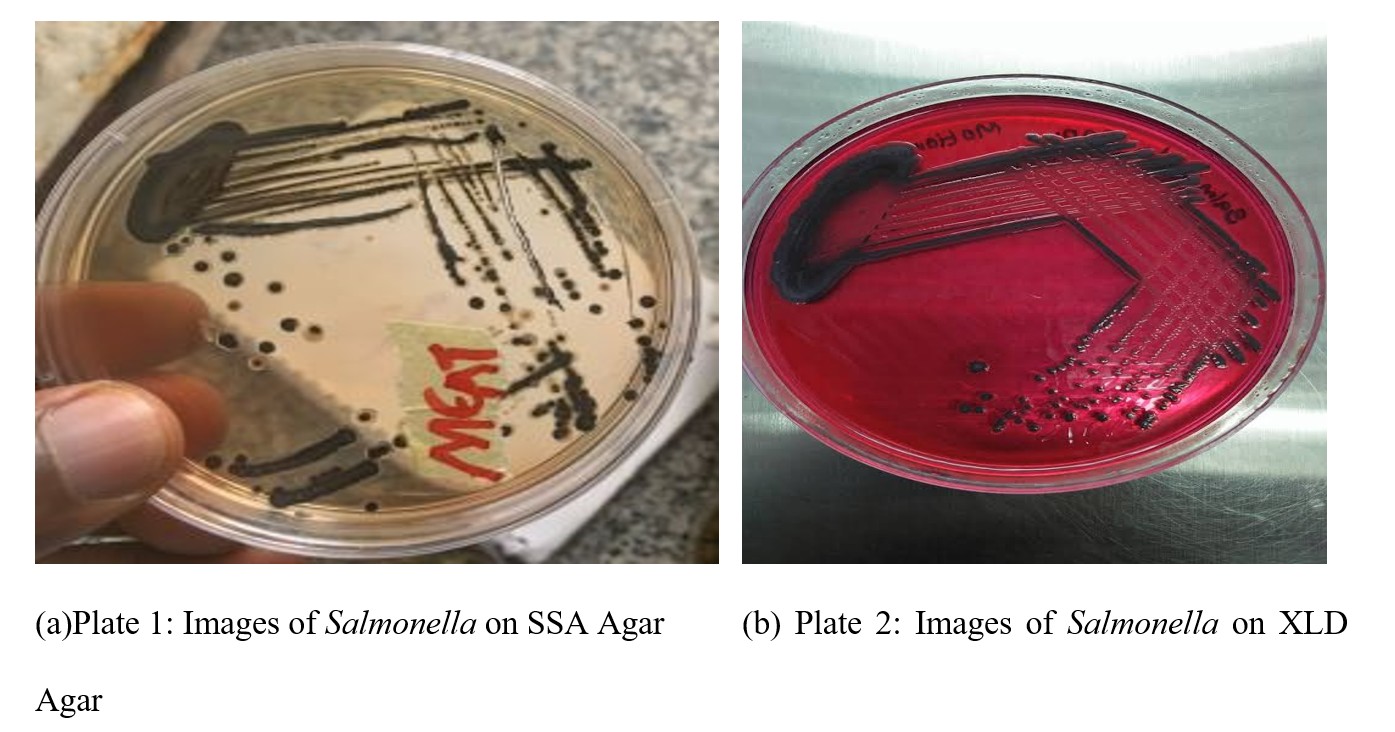
Eighty-four (84) poultry-derived samples (28 feces, chicken meat, and egg samples each) were collected from five major markets in Osogbo metropolis: Sasa, Oluode, Igbona, Alekuwodo and Oja-Oba. Samples were placed in sterile zip-lock bags, transported on ice and processed within 6 hours of collection. Each specimen was pre-enriched by suspending 1 g in 9mL of buffered peptone water (BPW) and incubating at 37oC for 18 - 24 hours. Thereafter, 0.1mL Aliquots were transferred into 10mL of Rappaport-Vassiliadis broth for selective enrichment at 42°C for 24 hours (Bawa et al., 2020). Loopfuls from the enriched broth were streaked on Salmonella-Shigella Agar (SSA) and Xylose Lysine Desoxycholate agar (XLD agar) and incubated at 37oC for 24 hours. Typical colonies (red or colourless with black-centers) were sub-cultured, stocked and identified biochemically the procedures of Oliveira et al. (2003) including catalase, oxidase, H2S production on TSI agar, fermentation tests, indole, motility test, methyl red and urease tests (Andrews et al., 2018).
Prevalence was calculated as:
Prevalence (P) = Number of positive isolates / Total samples collected x 100.
2.4 In vivo Experimental Design
2.4.1 Animal preparation
Forty (40) female albino mice aged four weeks were obtained from a certified breeder (high-sanitary-status farms from mothers serologically negative to Salmonella). The mice were acclimatized for two weeks under controlled laboratory conditions (25°C temperature; 60 to 65% humidity; 12 hours light/dark cycle). The animals were provided standard feed and water ad libitum and monitored daily for health and behavioural changes.
2.4.2 Grouping and treatments
Mice were randomly divided into 5 experimental groups (n = 8 per group):
- Group I: Negative control – received phosphate-buffered saline only;
- Group II: Positive control – challenged with Salmonella typhimurium only;
- Group III: Low probiotics dose (250 mg/Kg) + infection;
- Group IV: Medium probiotics dose (500 mg/Kg) + infection;
- Group V: High probiotics dose (750 mg/Kg) + infection.
Each mouse received oral gavage (0.2mL per dose) daily from Day 0 –14. Infection occurred on Day 7, all groups except Group I were orally inoculated with S. typhimurium suspension (0.2mL; 4 × 10³ CFU/mouse).
2.4.3 Clinical monitoring
Probiotic administration continued for another 7 days post-infection. Animals were observed daily for changes in activity, clinical symptoms, and feed intake. On Day 15, mice were humanely sacrificed, and blood, liver and kidney tissues were harvested for microbiological, and molecular analysis (Chen et al., 2023).
2.5 Probiotic Preparation
Commercial probiotic formulations containing Lactobacillus acidophilus ATCC 4356, and Bifidobacterium longum BB536 (Nature Field, USA) were used. The probiotic blend was freshly reconstituted in sterile normal saline prior to each administration. Probiotic and pathogen inoculant were prepared following Chen et al. (2023), ensuring viable counts of 4×103 CFU/mouse for the bacterial challenge.
2.6 Quantitative PCR (qPCR) Analysis
Quantitative real-time PCR (qPCR) were employed to detect and quantify the expression of the invA gene using synthesized primers (Forward primers: 5’- TTGTCACCGTGGTCCAGTTT-3’ and Reverse primer: 3’- TACCGGGCATACCATCCAGA-5’) virulence gene of S. typhimurium in liver and kidney tissues of infected mice. The GapA was used as the internal control to normalize expression levels (Siala et al., 2017). All reagents, methods and equipment were obtained from Applied Biosystems.
2.6.1 DNA extraction
Approximately, 100 mg of kidney and liver tissue was homogenized in 1 mL Tris EDTA (TE) buffer (10 Mm Tris-HCl, 1 mM EDTA, pH 8.0). A 520 μL aliquot was transferred into sterile microtubes, and 15 μL of 20% SDS with 3 μL of Proteinase K (20 mg/mL) was added. After incubation at 37 ºC for 1 hour, 100 μL of 5 M NaCl and 80 μL of a 10% CTAB solution in 0.7 M NaCl were added and votexed. The suspension was incubated for 10 minutes at 65ºC and cooled for 15 minutes. DNA was extracted with chloroform: isoamyl alcohol (24:1), precipitated with isopropanol (1: 0.6) at –20ºC overnight and pelleted by centrifugation. The DNA was washed with 70% ethanol, air-dried, and dissolved in 50 μL of TE buffer (Siala et al., 2017).
2.6.2 RNA treatment
To remove residual DNA, 20 ng of RNA was treated with NEB DNase 1 (M0303) according to manufacturer’s guidelines. Following incubation at 37°C for 10 minutes, DNase was inactivated by adding 1 µL of 0.5 M EDTA and heating at 75°C for 10 minutes. Treated samples were stored at -20oC until use (Siala et al., 2017).
2.6.3 Gene quantification
Expression levels of the invA virulence gene were quantified using Luna® Universal qPCR Master Mix Protocol (M3003). Reaction mixtures (20 µL) contained 10 µL master mix, 0.5 µL each of forward primer and reverse primer, 0.5 - 1 µL reverse transcriptase and 2 µL of RNA template.
Thermal cycling conditions were: Reverse transcriptase: 42oC for 10 minutes, initial denaturation: 95°C for 60 seconds, 40-45 cycles of denaturation at 95°C for 15 seconds, extension and plate reading at 60°C for 30 seconds, and Final extension: 72oC for 10 minutes.
Amplification was performed on a Bio-Rad CFX96 Real-Time PCR system, and specificity confirmed by melt-curve analysis. GapA served as the reference gene for normalization.
Expression changes were calculated using:
- ∆Ct = Ct of Target gene (Ctinv A) – Ct of the Reference gene (CtGap A)
- ∆∆Ct = ∆Ct (sample) - ∆Ct (calibrator)
- Fold change = 2–∆∆Ct
Positive detection threshold: Ct ≤ 35 (Heymans et al., 2018; Xue et al., 2025).


{kind=link}