Prevalence of K. pneumoniae associated with bovine mastitis
A total of 27 isolates were confirmed as K. pneumoniae using selective culture media, a series of biochemical tests and 16S rRNA gene sequencing. In biochemical assays, the isolates were positive for catalase, citrate utilization, and lactose fermentation, and negative for indole, oxidase, and methyl red identified as K. pneumoniae. Source-specific evaluation showed a higher prevalence in milk samples (27.1%, 19 isolates) compared to fecal samples (16.0%, 8 isolates). The prevalence was calculated directly from observed frequencies.
Antibiogram profile of the K. pneumoniae isolates
A total of 27 isolates were tested against fifteen antimicrobial agents, and all isolates demonstrated MDR, exhibiting resistance to 3 ≥ antibiotics (File S1). The highest resistance was recorded for doxycycline (DO) with 21/27 (77.8%) resistant isolates, followed by ampicillin (AMP) and tetracycline (TE), each showing 20/27 (74.1%) resistance. Moderate resistance frequencies of 18/27 (66.7%) were observed for ciprofloxacin (CIP), nalidixic acid (NA), and sulfonamides (S3). Resistance to chloramphenicol (C) and cefoxitin (FOX) was also substantial, observed in 17/27 (63.0%) isolates. Similarly, azithromycin (AZM) and ceftriaxone (CRO) displayed resistance in 16/27 (59.3%) isolates each, while nitrofurantoin (F), oxacillin (OX), and imipenem (IPM) exhibited resistance in 15/27 (55.6%) isolates. Moderate resistance was seen with Streptomycin (S) (14/27, 51.9%), whereas gentamicin (CN) showed the lowest resistance level (9/27, 33.3%) and the highest susceptibility (14/27, 51.9%) (File S1). Notably, three isolates (MBBL2, MNH_G2C5, and MNH_G2C5F) exhibited extensive multidrug resistance, with resistance to 13 of the 15 antibiotics tested. These three MDR isolates were selected for detailed genomic characterization, and their resistance profiles are summarized in Fig. 1.
Genomic features of K. pneumoniae strains
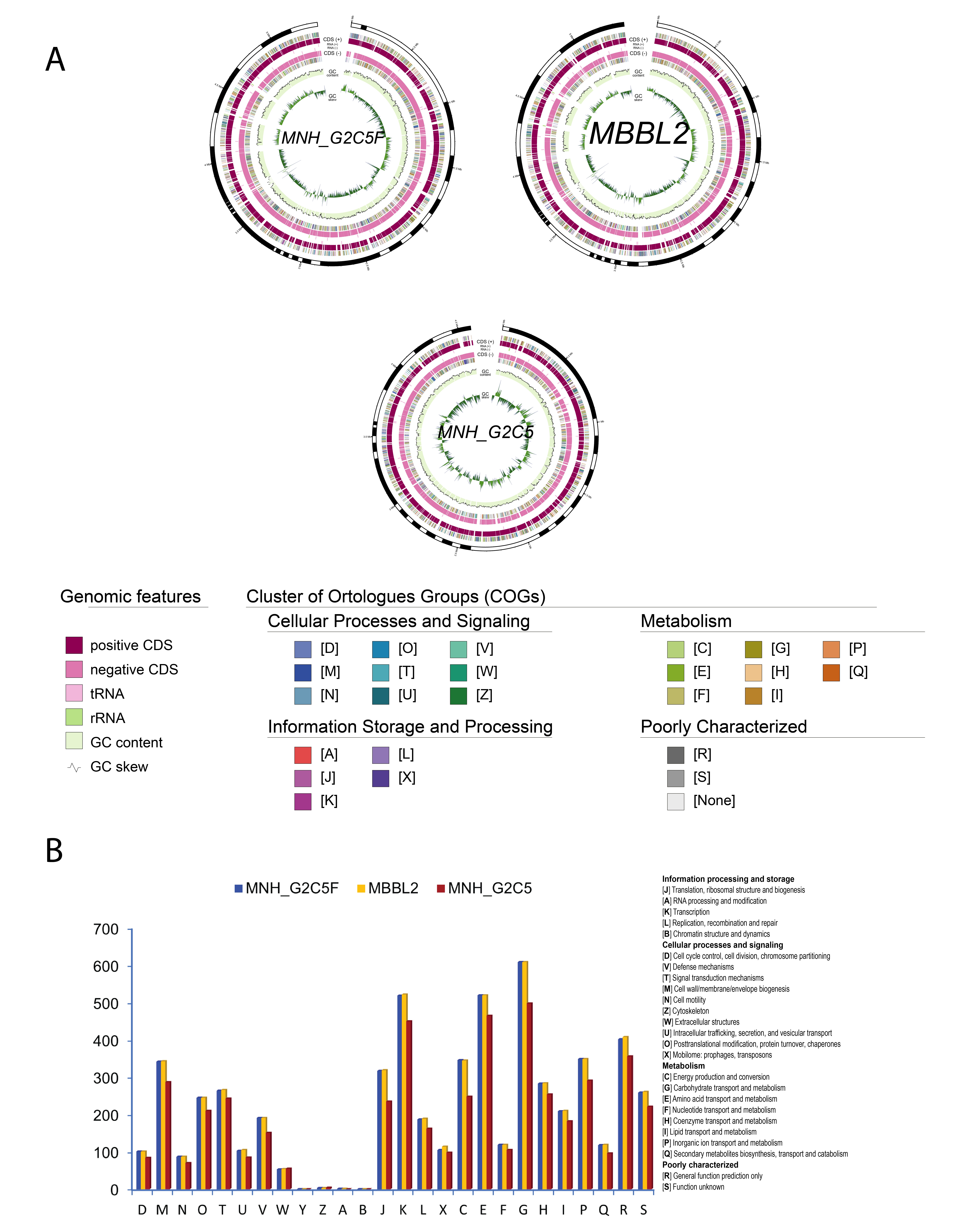
The assembled genomes of K. pneumoniae strains MBBL2, MNH_G2C5, and MNH_G2C5F were approximately 5.39 Mbp, 4.59 Mbp, and 5.38 Mbp in size, respectively, each with a GC content of 57.5%. Genome quality assessment indicated high completeness (> 98%) and low contamination (0.82%) for MBBL2 and MNH_G2C5F, whereas MNH_G2C5 exhibited reduced completeness (80.93%) and slightly elevated contamination (2.35%). We predicted 5,341 coding sequences in MBBL2, 4,478 in MNH_G2C5, and 5,311 in MNH_G2C5F. Additionally, the genomes contained 89, 63, and 87 RNA genes, respectively. Further genomic characterization revealed the presence of a single CRISPR array in MBBL2 and MNH_G2C5F, while no CRISPR elements were detected in MNH_G2C5. Functional subsystem classification assigned 393 subsystems to MBBL2, 341 to MNH_G2C5, and 395 to MNH_G2C5F. A detailed summary on the genomic features of these genomes is provided in Table S3.
The circular genome representations of K. pneumoniae strains MBBL2, MNH_G2C5, and MNH_G2C5F, together with their COG-based functional annotations, are shown in Figure S1A and S1B. COG classification assigned genes into 27 functional categories across four major groups. The most abundant category was G (carbohydrate transport and metabolism), comprising 10.3%, 10.0%, and 10.4% of the genes in MBBL2, MNH_G2C5, and MNH_G2C5F, respectively. This was followed by category E (amino acid transport and metabolism; 8.8–9.4%), category K (transcription; 8.9–9.0%), category R (general function prediction only; 6.9–7.2%), and category P (inorganic ion transport and metabolism; ~6%). Additional enriched categories included C (energy production and conversion; 5.0–5.9%), M (cell wall/membrane/envelope biogenesis; 5.8%), and J (translation, ribosomal structure, and biogenesis; 4.7–5.4%).
Molecular typing and genomic epidemiology of K. pneumoniae strains
To elucidate the global genomic epidemiology of K. pneumoniae, we reconstructed the phylogenetic relationships among three study isolates (MBBL2, MNH_G2C5, and MNH_G2C5F) and 591 global genomes from NCBI GenBank using a seven-gene MLST scheme (gapA, infB, mdh, pgi, phoE, rpoB, tonB) (Fig. 2). K. pneumoniae strains MBBL2 and MNH_G2C5F were assigned to sequence type 273 (ST273), differing by 97 core genome MLST (cgMLST) alleles, whereas MNH_G2C5 formed a distinct clade within ST101, separated by 747 alleles (Figs. 2A–C). cgMLST analysis revealed that MBBL2 and MNH_G2C5F clustered with 18 MDR ST273 strains from nine countries, while MNH_G2C5 showed close relatedness to 273 ST101 strains from 32 countries. All study strains exhibited genetic links to both human and animal-derived isolates (File S1). An SNP-based phylogeny, constructed from 50,138 core SNPs across 52 genomes (including 49 reference genomes), placed MBBL2, MNH_G2C5F, and MNH_G2C5 in a clade predominantly composed of human-derived strains, except for one environmental isolate (STIN_94). MBBL2 and MNH_G2C5F co-clustered with clinical strains KP_NORM_BLD_116392 and ARLG-3223, while MNH_G2C5 grouped with C17KP0052, a human bloodstream infection isolate (Fig. 3, File S2). This topology suggested recent common ancestry for MBBL2 and MNH_G2C5F, with MNH_G2C5 representing an earlier divergence.
Genomic diversity and strain-specific genes in K. pneumoniae strains
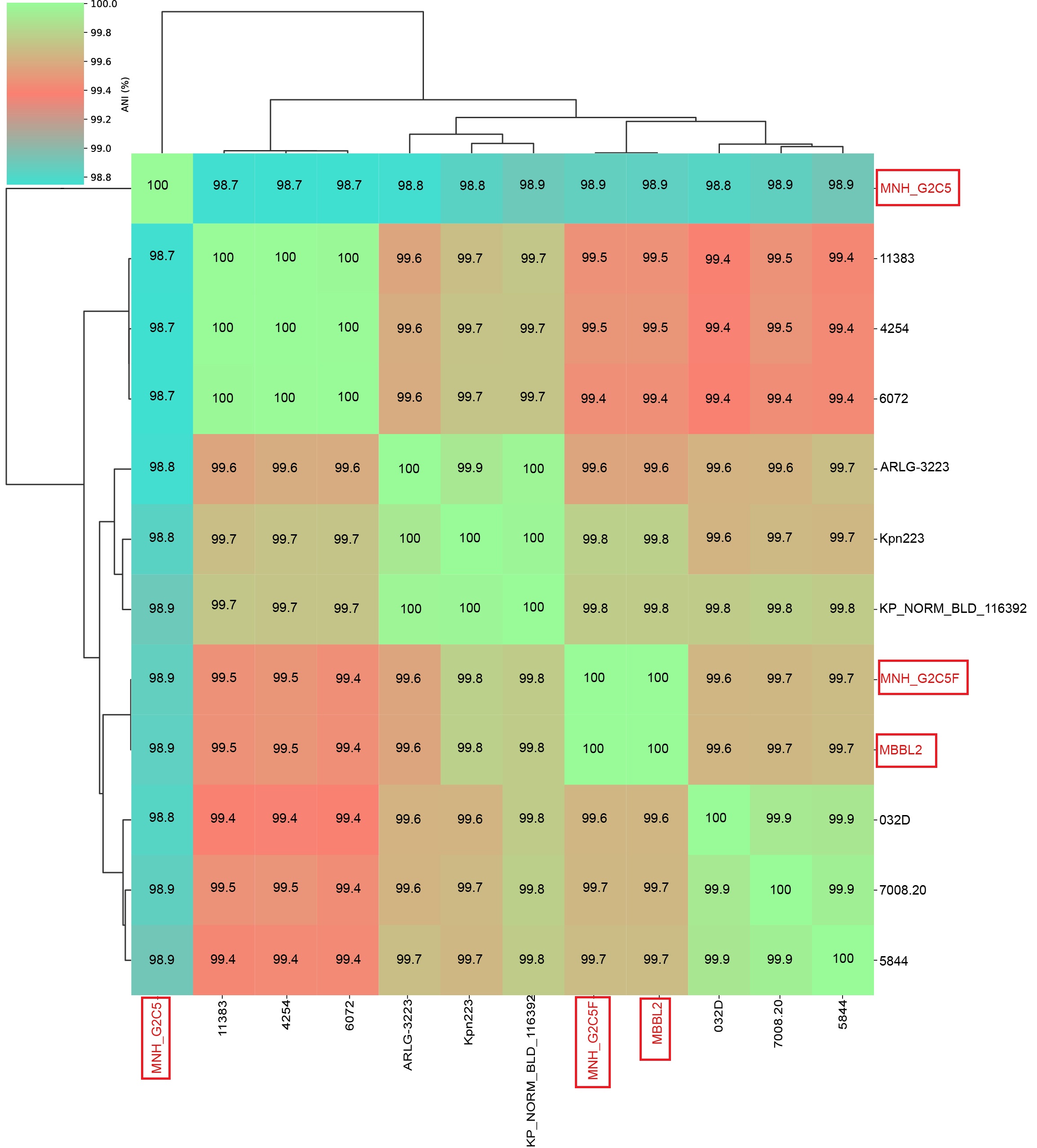
To substantiate the phylogenetic relationships and provide high-resolution genomic demarcation of the study isolates, we performed ANI analysis on a curated panel of 12 closely related strains (including study strains), selected based on their phylogenetic proximity from an initial set of 52 global genomes (Table S2, Figure S2). This analysis robustly confirmed the species-level identity of all three study strains (MBBL2, MNH_G2C5, and MNH_G2C5F), each demonstrating > 98% ANI with established K. pneumoniae reference strains, far exceeding the 95% threshold for species delineation [53]. The ANI results provided precise quantification of the genetic distances inferred from phylogeny. MBBL2 and MNH_G2C5F were nearly identical (99.9% ANI), whereas MNH_G2C5 showed slightly lower similarity (98.9%), reflecting its more distant phylogenetic position. MBBL2 and MNH_G2C5F exhibited the highest ANI (99.8%) with Kpn223 and KP_NORM_BLD_116392. In contrast, MNH_G2C5 was most closely related (98.9% ANI) to a broader set of strains, including ARLG-3223, Kpn223, KP_NORM_BLD_116392, MNH_G2C5F, MBBL2, 032D, 7008.20, and 5844 (Table S2, Figure S2).
A pangenome analysis was performed on the study genomes (MBBL2, MNH_G2C5, and MNH_G2C5F) together with nine reference genomes (Table S2) to explore their gene repertoire. The pangenome dendrogram revealed that MBBL2 and MNH_G2C5F were closely related to reference K. pneumoniae strains 5844, 032D, and 7008.20, while MNH_G2C5 showed the highest similarity to strains 6072, 11383, and 4254 (Fig. 4A). We identified 7,873 genes across the 12 K. pneumoniae genomes (Fig. 4B, Table S4). Strain-specific analysis further showed that MBBL2 possessed 1 unique gene and 826 accessory genes, MNH_G2C5 carried 123 unique genes and 234 accessory genes, whereas MNH_G2C5F, notably, contained no unique genes but shared 826 accessory genes (Fig. 4C, Table S4). The new gene accumulation curve (Fig. 4D) followed a power-law decay (y = 312.947x− 1.19, R² = 0.981963), indicating that the number of novel genes declines steadily with the addition of more genomes but does not reach a plateau, consistent with an open genome concept. Similarly, the pangenome expansion curve (Fig. 4E) also supported an open pangenome model, as the total number of gene clusters continued to rise with each additional genome (y = 200124x0, R² = 0.1), reflecting high genetic diversity. In contrast, the core genome displayed a progressive reduction, best fitted by an exponential decay model (y = 1358.75e− 0.08x + 2343.19, R² = 0.994518), suggesting gene loss with the inclusion of additional genomes.
Genomic plasticity and mobile genetic elements in K. pneumoniae strains
To assess the genomic plasticity of 12 phylogenetically related K. pneumoniae genomes, we employed a pangenome-based approach to predict regions of genomic plasticity (RGPs), defined as gene clusters located within highly variable genomic regions. The number of RGPs varied widely across genomes, ranging from 11 in KP_NORM_BLD_116392 to 34 in MNH_G2C5, which harbored the highest count. Notably, MBBL2 and MNH_G2C5F carried an equal number of RGPs (27 each) (Fig. 5). The sizes of these regions spanned from approximately 0.26 Mb to 1.061 Mb (File S3). To further explore contributors to genomic plasticity, we examined MGEs, including plasmids, prophages, and ISs. Plasmid profiling revealed four distinct replicon types among the three isolates. Both MBBL2 and MNH_G2C5F harbored IncFIB(K)(pCAV1099-114), IncFII(K), and IncHI1B(pNDM-MAR) plasmids while MNH_G2C5 carried only an IncR plasmid. Prophage counts varied across strains, with some harboring only intact prophages and others carrying a mixture of intact and questionable ones. MNH_G2C5 contained four prophage regions, whereas MBBL2 and MNH_G2C5F each carried two. Across the 12 K. pneumoniae genomes, a total of 816 IS elements were detected, of which 58, 52, and 46 were present in MBBL2, MNH_G2C5F, and MNH_G2C5, respectively (File S3).
Resistome repertoire in K. pneumoniae strains
Comprehensive resistome analysis of 12 closely related K. pneumoniae genomes highlighted the genomic diversity and adaptability of these strains to multiple stressors, including acid, AMR, biocides, and metals. The three study genomes viz. MNH_G2C5, MBBL2, and MNH_G2C5F carried 41, 30, and 29 resistome genes, respectively (Fig. 6, File S4), all of which included a strong core of acid resistance genes (e.g., asr). A total of 48 AMR genes, conferring resistance to 12 antibiotic classes, were identified across the three genomes. The AMR gene count was highest in MNH_G2C5F (18), followed by MBBL2 (17) and MNH_G2C5 (13). These genes conferred resistance to aminoglycosides (e.g., aac(3)-IId, aadA5), beta-lactams (e.g., blaCTX-M-15, blaLAP-2, blaSHV-1, blaSHV-11, blaTEM-1), trimethoprim (e.g., dfrA1, dfrA14, dfrA17), phenicols (e.g., oqxA, oqxB, oqxB20), tetracyclines (e.g., tet(A), tet(D)), sulfonamides (e.g., sul1, sul2), fluoroquinolones (e.g., qnrS1), fosfomycin (e.g., fosA), rifamycin (e.g., arr), macrolides (e.g., mph(A)), and efflux pumps (e.g., emrD), representing a shared resistance core (Fig. 6). The qacEdelta1 gene, conferring resistance to quaternary ammonium compounds, was detected in all three strains, suggesting tolerance to common chemical disinfectants. In addition, 34 metal resistance genes were detected in the three genomes. Arsenic resistance genes (e.g., arsC) were present in all strains, while mercury resistance genes (e.g., merA/C/P/R/T) were found in MBBL2 and MNH_G2C5F. Notably, copper resistance genes (e.g., pcoA/B/C/D/R/S) and silver resistance genes (e.g., silA/B/C/E/F/P/R/S) were exclusive to MNH_G2C5. Furthermore, MNH_G2C5 harbored nine heat resistance genes (e.g., psi-GI, kefB-GI, trxLHR, hdeD-GI, yfdX1/2, shsP, clpK, hsp20) (Fig. 6, File S4).
Virulence gene arsenal and mechanistic insights in K. pneumoniae strains
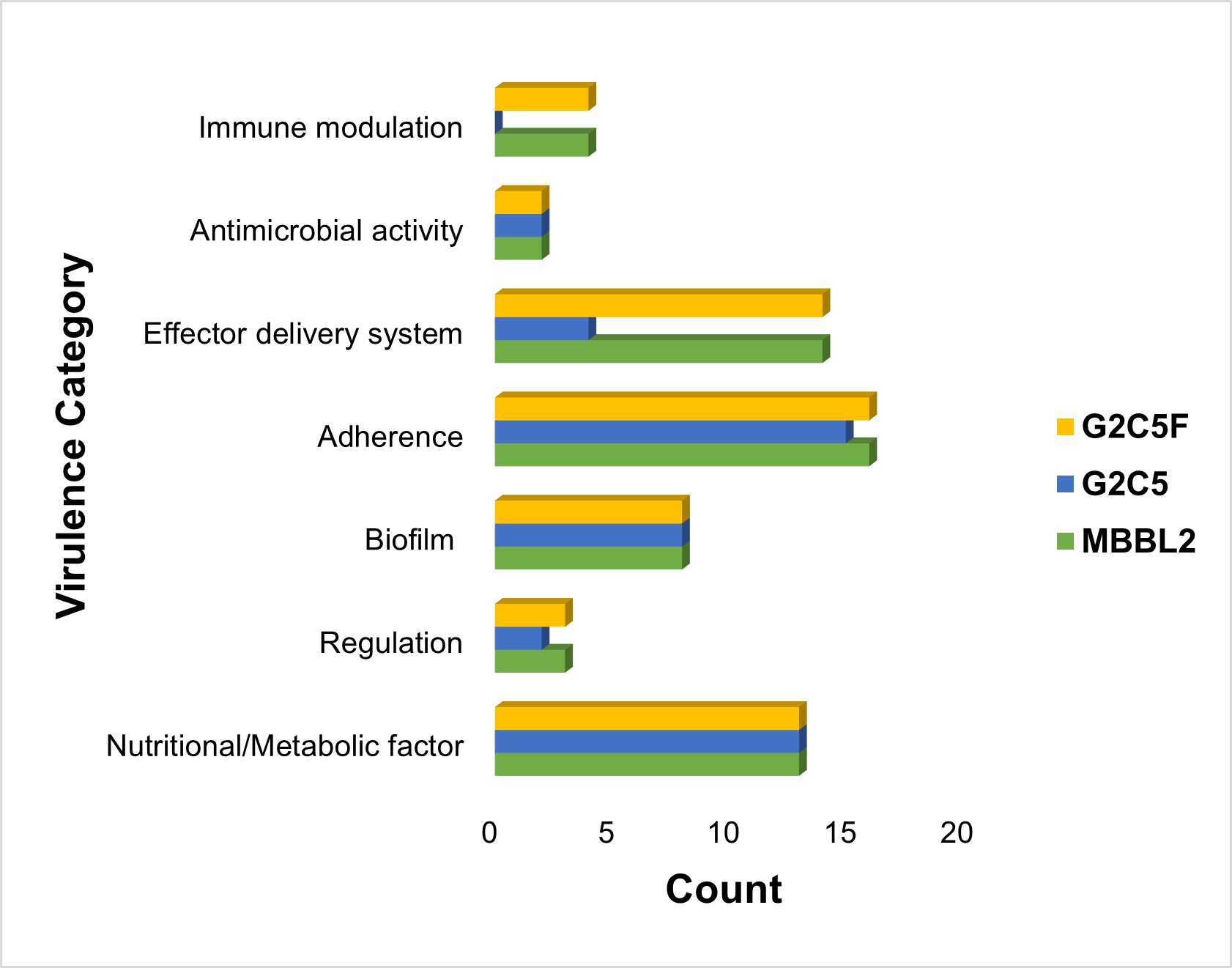
Virulence factor gene (VFG) profiles of the three study genomes were analyzed to assess their pathogenic potential. MBBL2 and MNH_G2C5F each harbored 60 VFGs, whereas MNH_G2C5 contained 44. These VFGs were found to facilitate virulence through several distinct mechanisms, including nutritional/metabolic factor, regulation, biofilm, adherence, effector delivery system, antimicrobial activity, immune modulation (Figure S3, File S4). In all genomes, adherence-related genes represented the largest category, accounting for 26.67% in MBBL2 and MNH_G2C5F and 34.1% in MNH_G2C5. Moreover, MBBL2 and MNH_G2C5F genomes possessed 14 effector delivery system genes (23.33%), 13 nutritional/metabolic genes (21.67%), eight biofilm-related genes (13.33%), four immune modulation genes (6.67%), three regulatory genes (5%), and two antimicrobial activity genes (3.33%). In contrast, MNH_G2C5 contained 13 effector delivery system genes (29.54%), eight biofilm-related genes (18.18%), four genes each for regulation and antimicrobial activity (4.54% each), and lacked immune modulation genes (Figure S3, File S4).
Prediction of genomic islands in K. pneumoniae strains
Genomic islands (GIs), which are gene clusters frequently acquired through horizontal gene transfer (HGT), were predicted in all three genomes and were found to harbor multiple VFGs and pathogen-associated genes, underscoring their potential role in pathogenicity. In the MBBL2 strain, 37 putative GIs were identified, of which 25 were predicted by SIGI-HMM and 12 by IslandPath-DIMOB, with sizes ranging from 4,035 to 291,727 bp. Among them, ten GIs carried genes associated with either virulence factors (VFs) or AMR. Notably, MBBL2 harbored curated AMR genes, including aac(3)-IId (WP_000557454.1), dfrA17 (WP_001389366.1), mrxA (WP_000004159.1), sul1 (WP_000259031.1), sul2 (WP_001043260.1), ACHL6M_RS25720 (WP_001516695.1), ACHL6M_RS25995 (WP_000503573.1), and ACHL6M_RS26045 (WP_000219391.1). In addition, homologs of resistance genes such as alaS (WP_004174700.1), floR (WP_023300759.1), kexD (WP_004200176.1), tetA (WP_000804064.1), ugd (WP_023328477.1), and ACHL6M_RS08150 (WP_023341379.1) were also identified (Fig. 7).
In comparison, the MNH_G2C5 strain harbored 38 GIs, with 25 predicted by the SIGI-HMM method and 13 by IslandPath-DIMOB, ranging from 3,010 bp to 46,746 bp in length. Four of these islands contained genes associated with VFs or AMR. MNH_G2C5 carried multiple curated resistance genes, such as PGZ78_RS07210 (WP_001516695.1), PGZ78_RS07200 (WP_000027057.1), dfrA1 (WP_000777554.1), and sul1 (WP_000259031.1), as well as homologs of resistance genes, including tetA (WP_000804064.1), and PGZ78_RS00380 (WP_032419057.1). Notably, one pathogen-associated gene, PGZ78_RS16620 (WP_000376623.1) was also identified (Fig. 7). Similarly, MNH_G2C5F contained 39 GIs, comprising 25 predicted by the SIGI-HMM method and 14 by IslandPath-DIMOB, spanning 4,035–190,576 bp, with ten GIs carrying VFs or AMR-related genes. This strain encoded multiple curated resistance genes such as Q2T98_RS25690 (WP_001516695.1), Q2T98_RS25965 (WP_000503573.1), dfrA17 (WP_001389366.1), Q2T98_RS26015 (WP_000219391.1), mrxA (WP_000004159.1), aac(3)-IId (WP_000557454.1), sul1 (WP_000259031.1), and sul2 (WP_001043260.1). It also harbored homologs of resistance genes, including alaS (WP_004174700.1), floR (WP_023300759.1), kexD (WP_004200176.1), tetA (WP_000804064.1), ugd (WP_023328477.1), and Q2T98_RS08145 (WP_023341379.1) (Fig. 7).
K. pneumoniae strains utilized diverse metabolic pathways to facilitate adaptation and survival
The systemic metabolic pathways of gene products in the MBBL2, MNH_G2C5, and MNH_G2C5F genomes were analyzed to better understand how these strains utilized nutrients, interacted with their environment, and influenced health and disease processes. A total of 3,524 genes (68.7%) from MBBL2, 2,973 genes (68.9%) from MNH_G2C5, and 3,514 genes (68.8%) from MNH_G2C5F were successfully annotated and mapped to KEGG pathways, spanning six major categories and forty-one subcategories (Figure S4). Among the major categories, metabolism-related genes were the most abundant (MBBL2 = 1,712; MNH_G2C5 = 1,492; MNH_G2C5F = 1,711), followed by those associated with environmental information processing, genetic information processing, cellular processes, human diseases, and organismal systems. Additionally, 58, 56, and 58 drug (antimicrobial) resistance genes were identified in MBBL2, MNH_G2C5, and MNH_G2C5F, respectively. All three genomes carried a complete biosynthesis of amino acids pathway that contributed to biogenic amine production. Seventeen complete modules related to production of biogenic amine identified in MNH_G2C5, while both MBBL2 and MNH_G2C5F genomes have 21 complete modules which correspond to serine biosynthesis, methionine biosynthesis, histidine biosynthesis and shikimate pathway respectively (Figure S5, File S5). Further analysis revealed that the three genomes harbored multiple genes influencing cellular community, including 71 quorum sensing genes in MBBL2 and MNH_G2C5F and 63 in MNH_G2C5 (e.g., luxS, qseC, qseB, lsrA, hfq, etc.), as well as biofilm-associated genes homologous to those characterized in Vibrio cholerae (e.g., cysE, wecB, csrA, etc.), Pseudomonas aeruginosa (e.g., gacS, crp, barA, etc.), and Escherichia coli (e.g., glgA, bcsA, rpoS, etc.) (File S5). In addition, both MBBL2 and MNH_G2C5F genomes possessed 45 beta-lactam resistance genes while MNH_G2C5 contained 44 beta-lactam resistance genes. All three genomes also carried five vancomycin resistance genes and five platinum drug resistance genes. Moreover, MBBL2 and MNH_G2C5F harbored 58 cationic antimicrobial peptide (CAMP) resistance genes and seven antifolate resistance genes, compared with 57 and five, respectively, in MNH_G2C5. Moreover, all three genomes possessed 19 genes (e.g., dxr, ACAT, atoB, ispA, uppS, ispE, ispD, dxs) associated with terpenoid backbone biosynthesis, along with 15 genes (e.g., entA, entB, dhbB, vibB, mxcF, entC, entD, pptT) related to the biosynthesis of siderophore group nonribosomal peptides.
Secondary metabolite gene clusters and human pathogenic potential in K. pneumoniae strains
The genomes of MBBL2, MNH_G2C5, and MNH_G2C5F harbored multiple biosynthetic gene clusters (BGCs) linked to secondary metabolites (Fig. 8). MBBL2 and MNH_G2C5 contained a redox-cofactor gene cluster, including pqqC, pqqD, and pqqE, involved in pyrroloquinoline quinone (PQQ) biosynthesis. Both genomes also encoded two azole-containing RiPPs (ribosomally synthesized and post-translationally modified peptides) encoded by pflA and ycaO (Figs. 8A and 8C). Moreover, MBBL2 and MNH_G2C5F genomes carried an NRP (non-ribosomal peptide)-metallophore gene cluster for enterobactin biosynthesis (entF, entC, entA) and a terpene-precursor which is encoded by ispA gene (Figs. 8A and 8B). Importantly, all of three genomes possessed a RiPP-like protein dsbA involved in disulfide bond formation (Figs. 8A-C). Additionally, BAGEL4 analysis identified rlmN (BmbF) in K. pneumoniae MNH_G2C5 (Protein ID: MDB1120801.1), a conserved bifunctional rRNA/tRNA methyltransferase experimentally validated at the protein level (PE = 3). The protein showed 100% sequence identity with characterized methyltransferases across multiple K. pneumoniae strains. Besides, MNH_G2C5 genome harbored BmbF (dual-specificity RNA methyltransferase RlmN) protein (Protein Id = MDB1120801.1) (Fig. 8D).
Simultaneously, the pathogenicity prediction analysis suggested that all three K. pneumoniae genomes possess a high likelihood of being human pathogens, as evidenced by PathogenFinder scores of 0.901 for MBBL2 and MNH_G2C5F, and 0.911 for MNH_G2C5. These values corresponded to the identification of 322, 255, and 322 pathogenic gene families across these genomes, respectively.
EPS and capsule biosynthesis gene clusters in K. pneumoniae strains
Further analysis of EPS and capsule biosynthesis clusters in three K. pneumoniae genomes revealed a largely conserved architecture with minor variations (Fig. 9). All genomes harbored two EPS/capsule biosynthesis clusters, with the MBBL2 and MNH_G2C5F clusters showing relative similarity. Both MBBL2-C1 and MNH_G2C5F_C1 contained a canonical EPS/capsule cluster, including genes involved in sugar-nucleotide biosynthesis (ugd, gndA), glycosyltransferases and tailoring enzymes (GT, GT2, GT4, wbaP), regulation and signaling (wzc, wzb, wcaJ, galF), and export/translocation (wza, Wzi, EpsG) (Table S5). Notably, MNH_G2C5F_C1 shared nearly all genes with MBBL2-C1, differing only by one or two hypothetical or accessory genes. Conversely, MBBL2-C2 and MNH_G2C5F_C2 contained a distinct EPS-like cluster comprising glycosyltransferases (wecA, wecF, wecG, rffC, rffM), polymerization and chain-length control genes (wzyE, wzzE), sugar-nucleotide biosynthesis genes (rffA, rffG, rfbA, wecB, wecC), and export-associated genes (wzxE). These clusters were highly conserved between the genomes, suggesting functional equivalence in polysaccharide assembly. Additionally, MNH_G2C5_C1 carried genes for regulation and signaling (rho), glycosyltransferases (wecA, wecF, rffM), polymerization/chain-length control (wzyE, wzzE), sugar-nucleotide biosynthesis (wecB, wecC, rffG, rfbA, rffA), and export (wzxE). MNH_G2C5_C2 featured a cluster including glycosyltransferases (GT, GT2, GT4, GT9, kbl, rfaC, rfaF, waaZ, waaA), accessory enzymes (tdh), sugar-nucleotide biosynthesis (rfaD), and export (waaL) (Fig. 9, Table S5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}