Cell Culture
Cell lines utilized in this manuscript were routinely authenticated with short tandem repeat profiling and Mycoplasma testing (Laboratory Corporation of America). Cells used for experiments that were grown in suspension were routinely replaced within two months after being revived from liquid nitrogen storage. Primary cells used for experiments were used within 10 passages after being revived from liquid nitrogen storage. Medulloblastoma cell lines (and their molecular sub-group) used in this study include: D425 (Group 3), MED-411FH (Group 3), MED-2112FH (Group 3), and CHLA-01-MED (Group 4). D425 Med (D425) cells (RRID: CVCL_1275) were derived from a primary medulloblastoma tumor resection at Duke University Medical Center and obtained from the laboratory of Dr. Jeffery Leonard[23, 24]. Med-411FH (MED411) cells and Med-2112FH (MED2112) cells are cell lines derived from early passage patient-derived xenografts developed by the laboratory of Dr. Jim Olson and distributed by the Brain Tumor Resource Lab at Seattle Children’s Hospital[25]. CHLA-01-MED was obtained from American Type Culture Collection as described previously[26]. Human meningeal cells (HMC) (ScienCell Research Laboratories, Cat. #1400) are primary leptomeningeal cells derived from human donors. We have used > 5 distinct lots with similar results. D425 and HMC cells were cultured in DMEM (Corning, 10-013-CV) supplemented with 10% FBS (Atlanta Biologicals, S11150H). MED411, MED2112, and CHLA-01-MED cells were cultured in NeuroCult NS-A Basal Medium (Human) (Stemcell technologies, Cat. #05750) supplemented with NeuroCult NS-A Proliferation Kit (Human) (Stemcell technologies, Cat. #05751), human epidermal growth factor (20 ng/mL, ThermoFisher, AF-100-15-500UG) and human fibroblast growth factor-basic (20ng/mL, ThermoFisher, 100-18B-50UG).
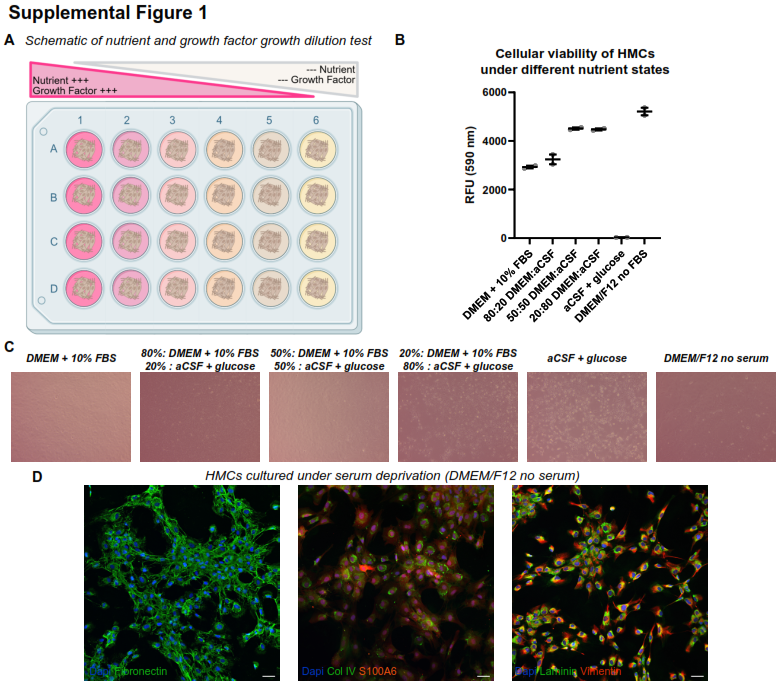
HMC survival optimization
HMC were grown to confluence in 24-well plates. Media was exchanged for either basal growth media, serial dilutions of growth media with artificial CSF (ACSF, Tocris, Cat. No. 3525), ACSF + 60mg/dL glucose, or DMEM/F12 (Gibco, Cat. #21041025). After 72 hours, cell viability was assessed using alamarBlue (ThermoFisher, A50100), and cells were photographed under light-phase microscopy.
Medulloblastoma-leptomeningeal fibroblast co-culture
Human medulloblastoma cells and meningeal cells were co-cultured to generate an in vitro model that captures key interactions between tumor cells and the surrounding stroma of leptomeningeal metastasis within a reductionist setting. HMCs were seeded onto 0.4 µm transwell filters or 1.5# glass coverslips (Electron Microscopy Sciences, 72204-04) in 6- or 24-well dishes and cultured with DMEM supplemented with 10% FBS. After HMCs formed a confluent monolayer (48–72 hours), the base media was removed and replaced with serum-free, growth-factor free DMEM/F12 containing medulloblastoma cells. For transwell experiments, DMEM/F12 was placed in the top and bottom wells.
Cell-free extracellular matrix
HMC were grown to confluence on 1.5# coverslips for 72 hours. Cell-free HMC-derived matrix was generated as previously described [27]. Medulloblastoma cells were plated onto coated coverslips for 48–72 hours in their base media before processing for immunofluorescence.
Animal Studies
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Abigail Wexner Research Institute at Nationwide Children’s Hospital (IACUC protocol AR15-00022). CB17-SCID mice [CB17SC scid−/−; RRID: IMSR_TAC:cb17sc] were used for all in vivo experiments.
Subcutaneous Tumor Model: Mice were anesthetized and injected subcutaneously in the flank with 5 × 10⁵ medulloblastoma cells, with or without 1 × 10⁶ HMCs. Tumor growth was monitored biweekly via caliper measurements. Mice were euthanized, and tumors were harvested upon a total tumor volume of 2000 mm³.
Spontaneous Leptomeningeal Metastasis Model: Mice were anesthetized with isoflurane and placed in a stereotaxic apparatus (Kopf Instruments, Model 940). Intracerebellar injection of 8 × 10⁴ medulloblastoma cells in 3 µL of Neurocult media was performed at 2mm lateral, 2mm posterior, and 2.5 mm deep relative to lambda. Post-operative care included analgesia and daily monitoring. At neurological endpoint (rapid weight loss and an enhanced body condition score of less than 9, and/or neurological clinical signs including head tilts, seizures, and/or log rolling), mice were euthanized and processed for histology.
Experimental Leptomeningeal Metastasis Model: Medulloblastoma cells (2.5 × 10⁴ in 3 µL Neurocult media) were injected into the cisterna magna under aseptic conditions. Post-operative care and endpoint monitoring mirrored the spontaneous model.
Histology
Mice were euthanized at defined endpoints, sternotomy was performed, and mice were perfused with 20 ml of 0.9% saline injection (BD PosiFlush, cat #306546) followed by 30 ml of neutral buffered formalin (NBF) (Sigma-Aldrich, HT5012-1CS) through the left ventricle. Heads and spinal columns were cleaned of connective tissue and placed in NBF at 4°C for 48 hours. Brains were dissected from skull and spines were decalcified in 10% EDTA (Elabscience, E-IR-R112) for 10 days at room temperature, and then processed for paraffin embedding (FFPE). For standard histological observation, FFPE sections were counterstained with hematoxylin and eosin via conventional methods.
Immunohistochemistry and Immunofluorescence
Unstained 4µm sections were deparaffinized with xylene and rehydrated through ethanol series. Heat-mediated antigen retrieval was performed with Tris-EDTA buffer (TE; pH 9) with 0.2% v/v Tween 20 (Fisher Bioreagents, BP337-100).
Cells growing on #1.5 glass coverslips or 0.4µm filters were fixed in NBF for 10 minutes at room temperature. Following three rinses in 1x Phosphate Buffered Saline (PBS) (Corning, 21-040-CM), coverslip or histological sections were then incubated for at least 1 hour in blocking solution consisting of PBS, 0.2% triton100 (v/v) (Sigma, X-100-100ml), and 3% bovine serum albumin (w/v) (Sigma, A-7888) at room temperature. Coverslips or histological sections were then incubated overnight in the following primary antibodies (diluted in blocking solution): hamster anti-Podoplanin (Developmental Studies Hybridoma Bank(DSHB), 8.1.1), rabbit anti-Fibronectin (Abcam, ab2413), rabbit anti-Collagen IV (Abcam, ab6586), rabbit anti-Synaptophysin (Abcam, ab32127), rabbit anti-GFAP (Abcam, ab68428), rabbit anti-CD31 (Abcam, ab182981), rabbit anti-pan laminin (Novus Biologicals, NB300-144), rabbit anti- Cleaved Caspase 3 (Cell Signaling Technologies-CST, 9664), rabbit anti- phospho-Histone Ser10 (CST, 53348), goat anti-CD45 (R&D Systems, AF114), goat anti-OTX2 (R&D Systems, AF1979), sheep anti-S100A6 (R&D Systems, AF4584), or mouse anti-Vimentin (Abnova, SRL33). Following three rinses in PBS, sections or coverslips were incubated with appropriate AlexaFluor-labeled secondary antibodies (Invitrogen), DAPI (Invitrogen, D1306) and, where indicated, Phalloidin AlexaFluor 488, 568 or 647 (Invitrogen), or Wheat Germ Agglutinin (WGA) AlexaFluor 647 (Invitrogen, W11261) diluted in blocking solution for 1 hour at room temperature. Following PBS rinses, coverslips and tissue sections were mounted in Fluoromount G (Invitrogen, 00495802).
Microscopy and image analysis
Confocal microscopy images were obtained using a Crest-X-Light V3 spinning disk confocal on a Nikon Ti2-E inverted microscope, equipped with either a Hamamatsu ORCA Fusion camera or a Zeiss 800 laser scanning microscope, and featuring 4X, 20X, and 40X air objectives. Raw ND2 or czi files were imported into ImageJ for post-image processing and quantitation. All manipulation of images (brightness/contrast) were done uniformly to images. Mitotic (pH3+) and apoptotic (CC3+) were quantified by creating binary images with a manual threshold, then subjecting the binary images to the Analyze Particles function (size > 10µm2). Index was generated by dividing the cells (pH3 + or CC3+) by the total number of medulloblastoma cells (OTX2+). Spreading of medulloblastoma cells in co-culture was quantified by generating a binary image of OTX2 image and processed using the Skeletonize function. Skeletonized images were then quantified with Analyze Skeleton function. Percent spreading on HMC-derived ECM was quantified manually using the Cell Counter Plugin. H/E images were obtained with Aperio FL ScanScope digital slide scanner with 20X objective, and metastasis length was quantified using Aperio ImageScope Software (V12.4.3).
Statistical analysis
All statistical analyses for in vitro assays and animal studies were performed using GraphPad Prism v10. The number of biological replicates (independent experiments or animals) is specified in the figure legends, along with the appropriate statistical tests and P values. Data are presented as the mean with error bars that represent the standard error of the mean.
{kind=link}