Increased immunoreactivity of PQC factor VAPB inversely correlates with the absence of pathogenic aggregates in the MNs of multiple ALS subtypes.
Endoplasmic reticulum (ER) chaperones play critical roles in regulating proteotoxic effects and autophagy overload due to the accumulation of misfolded protein aggregates in ALS[39]. Consistent with the role of ER chaperones in providing resilience against toxic proteins[15, 16, 22], we recently demonstrated that elevated levels of VAPB were found in AD transgenic (Tg) mouse models including APP/PS1 and pR5 Tau (Tg) mice [114]. Similarly, AD patients' neurons affected by pathological phosphorylated tau (pTau) and granulovacuolar degeneration (GVD) showed differential immunoreactivity of VAPB, supporting the assumption that VAPB plays a role in maintaining neuronal PQC [114]. Thus, we hypothesized that VAPB supports selective neuronal survival by enhancing the autophagic clearance of toxic aggregates and that abnormal VAPB accumulation disrupts these protective mechanisms, contributing to neuronal vulnerability in ALS.
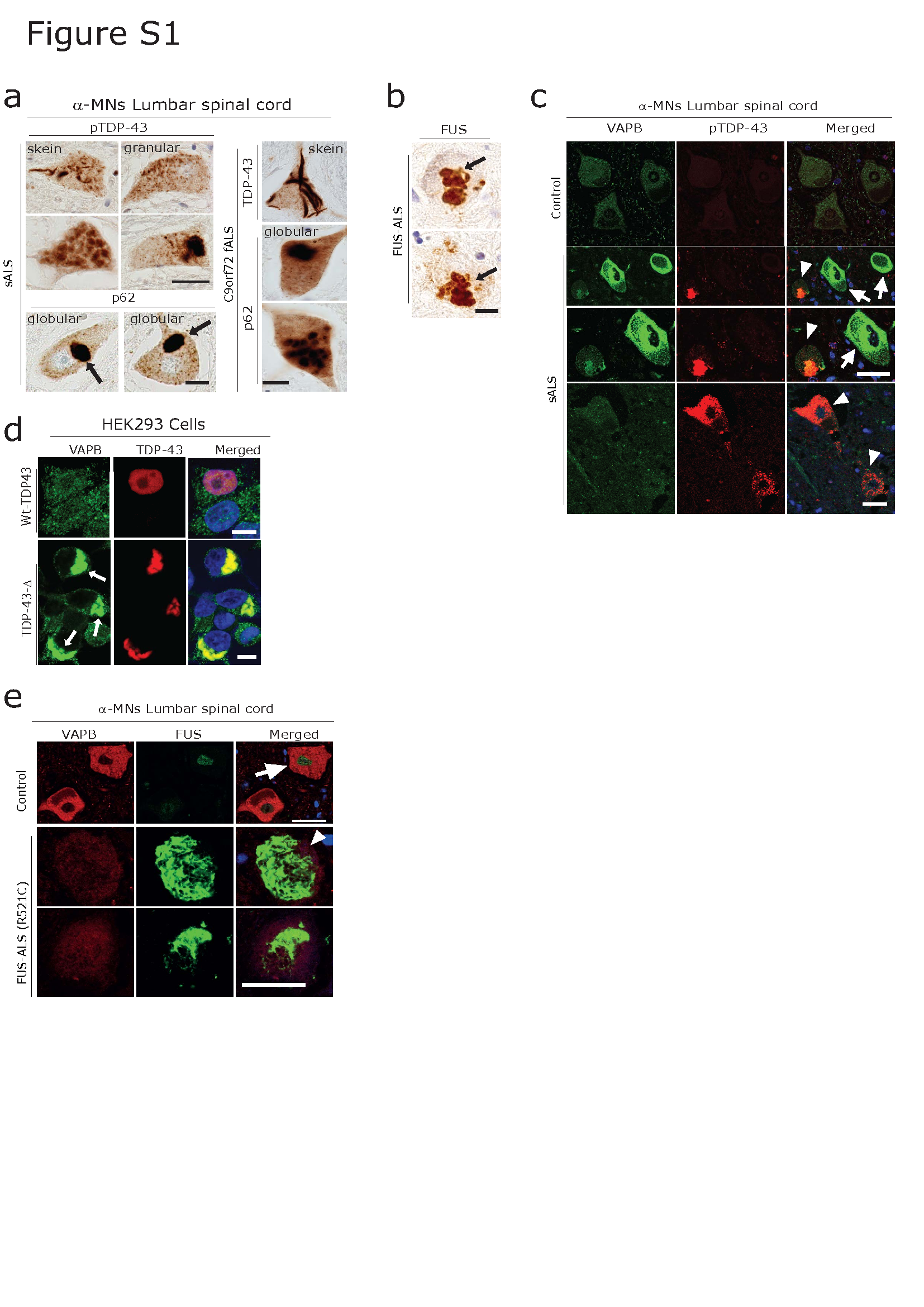
We focused on large diameter MNs in the lumbar spinal cord ventral horn as (putative) α-MNs which were recognized by their size (50-100µM) as well as markers including vesicular acetylcholine transporter (vAChT) [24]. We confirmed the presence of VAPB immunoreactivity in these large VAChT-positive putative α-MNs (yellow arrowheads) in human lumbar spinal cord (Figure 1a, b, c). These α-MNs frequently harbour pathologically phosphorylated TDP-43 (pTDP-43) and p62 aggregates in sporadic (s), as well as of C9orf72 familial (f) ALS (Figure 1a, b, quantification d, e, Figure S1a,). In FUS fALS, α-MNs are pTDP-43-negative and FUS aggregate-positive (Figure S1b).
Consistent with our hypothesis and along with our previous observation on TDP-43 proteinopathies and the role of ER chaperones[44, 87, 109], we found increased VAPB immunoreactivity associated with the ER in human ALS lumbar spinal cord α-MNs (Figure 1, Figure S1c) as well as in cortical neurons (Figure S3a) compared to the controls. α-MNs displaying increased immunoreactivity for VAPB were often devoid of p62 and pTDP-43 aggregates both in sALS as well as in C9orf72 fALS cases (Figure 1a and b, arrows, Figure S1c). In contrast, surviving α-MNs already harboring p62 and/or pTDP-43 aggregates displayed significantly reduced immunoreactivity of VAPB (Figure 1a, b, Figure S1c, arrowheads, quantification: Figure 1 d-e). Furthermore, we tested the pattern of VAPB immunoreactivity in lumbar spinal cord sections of FUS fALS patients. Again, α-MNs showing increased VAPB immunoreactivity were often devoid of FUS aggregates (Figure 1c). In contrast, surviving MNs harboring FUS aggregates displayed significantly reduced levels of VAPB (Figure 1c, Figure S1c, arrowhead, quantification d, e).
Dipeptide repeat (DPR) aggregates including poly-GA and poly-GR, together with pTDP-43 aggregates were abundant in cortical and hippocampal neurons and are central to the pathogenesis in C9orf72 fALS patients' brains [33, 60] (Figure 1f). In line with the VAPB immunoreactivity in α-MNs, we found similar differential immunoreactivity, where increased levels of VAPB negatively correlated with the absence of pTDP-43 and poly-GA aggregates, for example in C9orf72 fALS hippocampal neurons (Figure 1g).
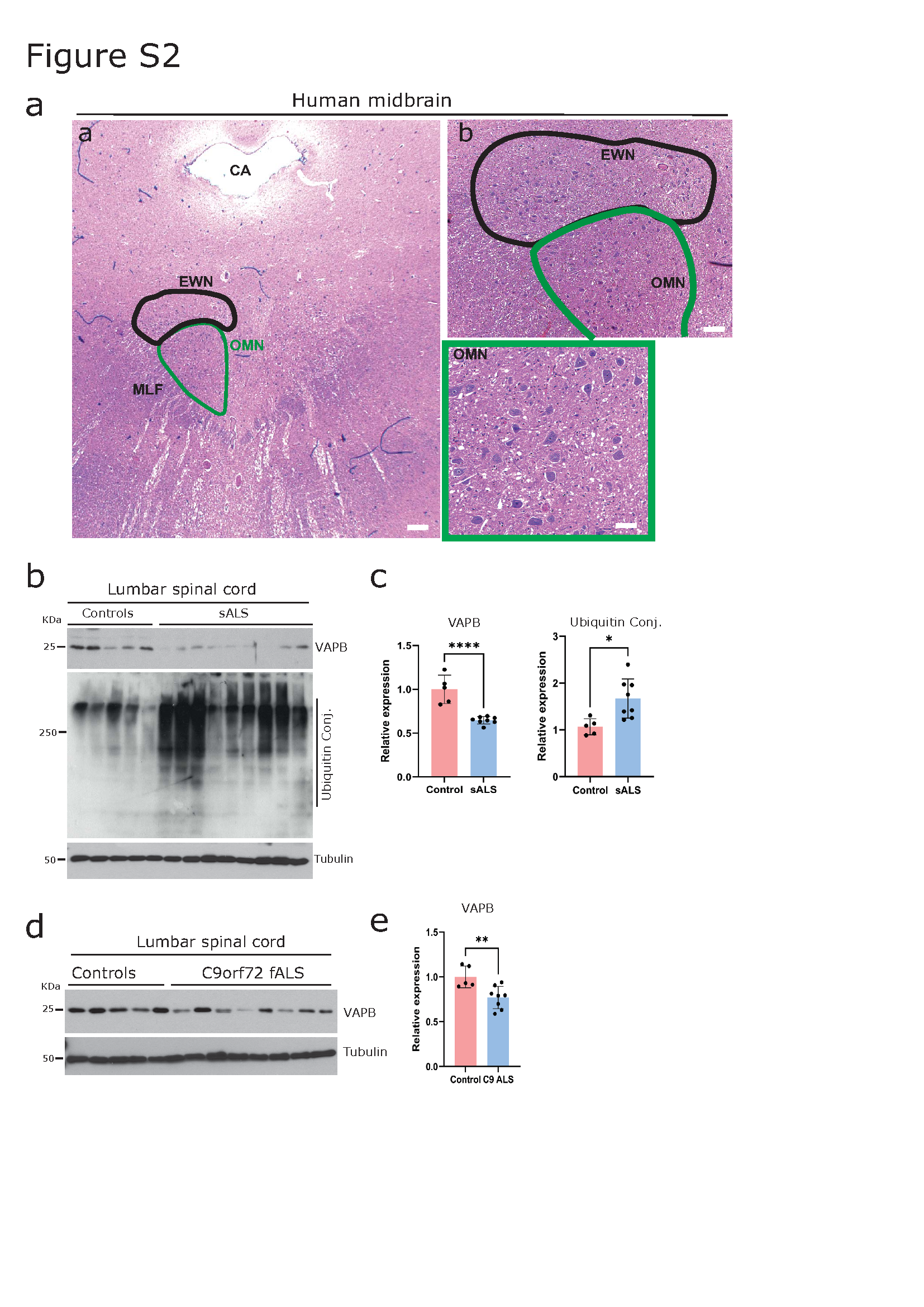
Immunoblot analysis performed on the lumbar spinal cord lysates obtained from sALS and familial C9orf72 fALS patients showed a clear decrease in VAPB protein levels (Figure S2 b-e). Together tthese results suggest that VAPB might exert selective neuroprotection by clearing pathogenic aggregates from the unaffected population of MNs in multiple subtypes of ALS.
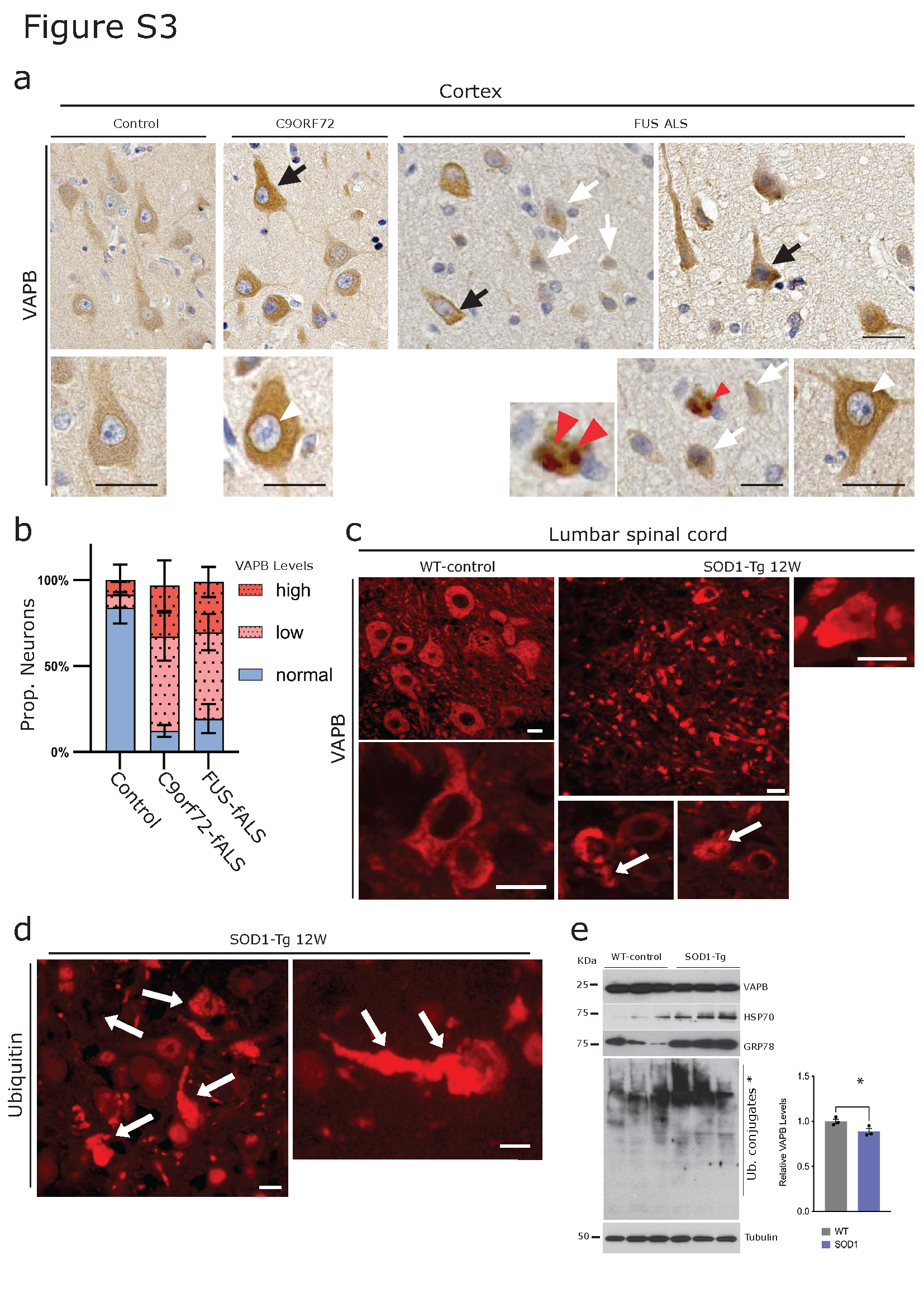
In line with the results obtained above from the lumbar spinal cord, DAB immunohistochemistry performed on C9orf72- and FUS-ALS primary motor cortex showed differential cytoplasmic immunoreactivity, with many neurons showing reduced VAPB immunoreactivity (Figure S3a, white arrows) while a few neurons showed increased cytoplasmic immunoreactivity and VAPB accumulation (Figure S3a, black arrows, and red arrowhead respectively). In many instances, a peculiar, intense nuclear envelope immunoreactivity was also observed in several pyramidal neurons in the C9orf72 fALS and FUS-ALS motor cortex (Figure S3a, white arrowheads).
In parallel, similar patterns of VAPB immunoreactivity to those observed in human ALS were also found in the SOD1 mouse model of ALS. VAPB immunoreactive aggregates as well as ubiquitin-positive aggregates were also evident in the MNs of lumbar spinal cords of SOD1 mice (Figure S3c-d). Overall, VAPB protein levels were reduced, as detected by IF analysis, accompanied by increased proteotoxicity as evidenced by elevated ubiquitin accumulation (Figure S3d). Consistently, WB analysis revealed altered levels of the chaperones GRP78 and HSP70, along with increased ubiquitin levels, in the lumbar spinal cord of SOD1 mice (Figure S3e; quantification shown).
ALS-resistant MNs display high levels of VAPB and are mostly devoid of pathogenic aggregates.
As ALS progresses, specific sub-types of MNs preferentially deteriorate while others are spared until the disease's end stage. For instance, MNs of Onuf’s nucleus in the sacral spinal cord and oculomotor nucleus in the midbrain exhibit resistance and are preserved in ALS [63, 82, 83]. Building upon this observation and our previous findings [44, 87], we investigated whether these disease-resistant MNs also display increased immunoreactivity for VAPB. We chose to analyze midbrain oculomotor neurons (Figure S2a) because they are more easily accessible compared to MNs of the Onuf’s nucleus of the sacral spinal cord [63, 82, 83]. VAPB shows a Nissl-associated pattern of immunoreactivity in the MNs (Figure 2) [44, 87, 109]. Consistent with the role of VAPB as a PQC factor, we observed a significantly elevated level of VAPB immunoreactivity in oculomotor neurons across various ALS sub-types when compared to controls (Figure 2a, quantification d). For comparison, we utilized the known endoplasmic reticulum (ER) chaperone GRP78, a key player in the PQC mechanism that exerts neuroprotective effects by reducing levels of misfolded proteins [37]. As expected, we found increased levels of GRP78, which co-localized with VAPB (Figure 2a).
We then investigated whether the elevated levels of VAPB correspond to reduced levels of aggregates in these MNs. Oculomotor neurons from both sALS and C9orf72 fALS appeared normal with no visible signs of atrophy or degeneration. They also exhibited increased levels of VAPB staining compared to control MNs. These MNs were largely devoid of any pathogenic pTDP-43 aggregates (Figure 2b). In only a few cases we were able to observe occasional p62 or pTDP-43 immunoreactive profiles in these neurons (not shown). Similarly, FUS aggregates were also very rare in FUS-ALS oculomotor neurons (Figure 2c, quantification e), and consistent with other sub-types of ALS, oculomotor neurons in FUS ALS displayed increased levels of VAPB. The high levels of VAPB in this population of MNs is consistent with the notion that it confers neuroprotection by preventing toxic aggregate formation.
VAPB is sequestered with pathogenic aggregates in ALS: failure of PQC?
While the PQC mechanism typically maintains neuronal health during stress, protein misfolding, aggregation and persistent proteotoxic stress caused by an increased amount of misfolded protein aggregates can compromise the PQC mechanism[1, 35, 107]. This results in a decline in neuronal proteostasis and further contributes to neurodegeneration[35, 102, 107]. In line with this idea, we observed aggregated unfolded protein response (UPR) factor VAPB in the lumbar spinal cord α-MNs of human sALS (Figure 3a). These aggregates exhibited various morphologies, reminiscent of the well-known pTDP-43 and p62-immunoreactive cytoplasmic neuronal aggregates in ALS (Figure S1a). To confirm the accumulated/aggregated VAPB, we performed the FTA with frozen lumbar spinal cord tissues from sALS and control patients. FTA analysis confirmed the presence of SDS-insoluble VAPB aggregates (Figure 3b, quantification right). To gain further insights into the morphology of these SDS-insoluble VAPB aggregates in the α-MNs, we conducted co-immunolabelling experiments of VAPB along with pTDP-43 and p62, which are the known pathological hallmarks in the α-MNs of human sALS [76]. Consistent with earlier reports [9], we observed that pTDP-43 and p62 aggregates exhibited diverse morphologies, including dash-like, skein-like, and granular/globular-like forms (Figure S1), and were co-localized with accumulated VAPB (Figure 3c, d; quantification in e) in the lumbar spinal cord α-MNs from sALS and C9orf72 fALS cases. Interestingly VAPB immunolabelling performed on HEK293 cells overexpressing the DPR (expanded poly GA) or mutant TDP43 showed the aggregation (arrow) and sequestration of endogenous VAPB together with the aggregates of poly GA and mutant TDP-43 (Figures 3f and Figure S1d respectively). These findings were confirmed in affected cortical (not shown) and hippocampal neurons of C9orf72 fALS cases, where aggregated DPRs (poly-GA) and pTDP-43 co-localized with VAPB (Figure 3g, h). Additionally, through biochemical analysis using FTA, we confirmed that VAPB forms SDS-insoluble aggregates in the lumbar spinal cord of C9orf72-fALS cases (Figure 3i and quantification), like that observed in sALS cases. Furthermore, the presence of aggregated VAPB was in line with the observation that soluble levels of VAPB were significantly decreased in both sALS and C9orf72 fALS, lumbar spinal cord lysates, as detected by Western blot analysis (Figure S2b-e, quantification).
VAPB is sequestered in FUS-ALS
Our next objective was to analyze whether VAPB plays a similar role in FUS-ALS, particularly in cases with FUS-R521C or P525L mutations, which cause a rare, rapidly progressive and severe form of ALS. Interestingly, VAPB immunolabelling performed on HEK293 cells stably expressing mutant FUS showed the aggregation and sequestration of endogenous VAPB (see quantification (Table S3) together with the aggregates of mutant FUS (arrows, Figure 4a). Consistent with this and with the previous observations (Figure 3), lumbar spinal cord α-MNs from FUS-R521C cases also exhibited the accumulation of VAPB along with its sequestration with FUS aggregates (Figure 4b, quantification right).
We then extended our investigation to FUS-ALS iPSC-derived MNs. These iPSC MNs have been demonstrated to faithfully represent FUS-ALS pathologies and exhibit age-dependent FUS aggregation (Figure 4c [75, 106]). Immunolabelling using VAPB antibody showed significant aggregation of VAPB in FUS mutant iPSC-derived MNs, whereas iPSC-derived control MNs showed a normal distribution of VAPB (Figure 4d). Interestingly, FUS aggregates were also found to be co-localized with VAPB aggregates (Figure 4d, see semi quantitative analysis (Table S3), consistent with the VAPB sequestration with FUS aggregates observed in FUS-ALS lumbar spinal cord MNs.
Aggregates of RBPs, including FUS, often proceed via the stress granule (SG) pathway. FUS has also been reported as a component of SG [66, 86, 111]. In addition, T cell intracellular antigen-1 (TIA-1 or Tia1) is a prion-related RNA-binding protein that is a well-known key component of SGs [61, 66]. In line with this, we also observed increased accumulations of Tia1-immunoreactive SGs (Figure 4e). Intriguingly, these Tia1-positive SGs also sequester VAPB. See semi quantitative analysis (Table S3).
VAPB is localized at the enlarged C-terminals of the lumbar spinal cord α-MNs in ALS
While examining VAPB staining in lumbar spinal cord α-MNs, we noticed a distinct VAPB signal at a specific type of synapse called the C-bouton (Figure 5a, white arrowheads). Earlier studies, including ours, have shown that C-bouton synapses become enlarged in ALS motor neurons, likely as a response to ongoing neuron loss. These synapses can be identified using VAChT staining (Figure 5a, yellow arrowheads). Interestingly, in ALS patient lumbar spinal cord α-MNs, VAPB staining was also present at these enlarged C-bouton synapses (Figure 5a)[89]. The enlarged C-bouton synapse is associated with several pre - and post-synaptic signaling proteins, including the ER chaperone SigR1, which accumulates at the post-synaptic side (Figure 5b, white arrowheads). Consistent with this, we observed that VAPB co-localized with SigR1 at these sites (Figure 5c, white arrowheads) [87]. Furthermore, VAPB immunoreactivity at the synaptic sites was confirmed by the presence of VAPB in the vicinity of postsynaptic Kv2.1 (Figure 5 d, white arrowheads). Finally, the presence of VAPB at the synaptic sites was confirmed using WB analysis from the synaptic fraction purified from the human lumbar spinal cord (Figure 5e).
Endogenous VAPB is a substrate for autophagy and the ubiquitin proteasome system.
VAPB was found to be aggregated in the ALS autopsy tissues and IPSC-derived MNs obtained from FUS-ALS patients. This phenomenon is intriguing because endogenous soluble VAPB levels are reduced in sALS and C9orf72 fALS lumbar spinal cord lysates (Figure S2 b-d) and FTA analysis in these samples showed increased SDS resistant insoluble VAPB aggregates (Figure 3b and i). Previous studies have demonstrated that both loss and toxic gain of mutant VAPB functions are associated with neurodegeneration [45, 50, 105] with disturbed autophagy being central to the pathogenesis[28, 52]. We therefore investigated a) whether inhibition of autophagy could lead to the accumulation of VAPB, or vice versa, and b) whether an accumulation of VAPB in any given instance are due to the failure of autophagy. To confirm this hypothesis, we treated cells with a known autophagy inhibitor Bafilomycin A (Baf.A) or with a proteasome inhibitor MG132 and then checked the level of VAPB under these conditions. As expected, blocking autophagy led to the increased accumulation of VAPB in HEK293 cells compared to MG132 treated cells (Figure 6a). Co-immunolabelling of VABP with either ubiquitin antibody or the ER stress marker GRP78 confirmed the proteotoxicity and increased ER stress upon Baf.A treatment (Figure 6b-d, quantification). Consistent with the findings, immunoblot analysis of HEK293 cells treated with either Baf.A or the ER stressor thapsigargin showed increased levels of VAPB. Besides, increased levels of ubiquitin and GRP78 and GADD-153 protein further confirmed the ongoing proteotoxic effect upon these inhibitors (Figure 6 e, f, quantification). In summary, these results indicate that VAPB is a substrate of autophagy and its accumulation/aggregation in cells/neurons indicates disturbed autophagy.
VAPB regulates autophagy: Increased turnover of autophagy substrates by controlled over-expression of VAPB
The present study observed that VAPB interacts with the autophagy protein p62 (Figure 3d) and that VAPB levels increase when autophagy is inhibited (Figure 6) and we also proposed that VAPB confers reduced vulnerability to toxic aggregates across multiple ALS sub-types (Figures 1 and 2). VAPB interacts with autophagy related (ATG) proteins to maintain endoplasmic reticulum (ER) and the isolation membrane (IM), ER/IM contacts which are essential for autophagosome biogenesis. However, considering the deleterious impact of the P56S-VAPB mutation on autophagosome biogenesis and late autophagy stages [109, 115], we hypothesize that increasing the levels of wild-type (Wt) VAPB can enhance autophagic flux. Supporting this hypothesis, we demonstrated that VAPB over-expression effectively degrades p62 bodies by inducing autophagy (Figure 7a, quantification 7d). Additionally, we observed reduced LAMP1 immunoreactivity, indicating enhanced turnover of LAMP1 due to increased autophagy activity (Figure 7b, quantification 7d). These results were consistent with our previous findings [44, 109] suggesting the role of VAPB in managing autophagy.
To further confirm that VAPB protein facilitated the induction of autophagy, we used Baf. A to block autophagy in cells overexpressing either control EGFP-, or EGFP-VAPB (Figure 7c) or control mCherry or VAPB-mCherry and monitored the protein levels of LC3 as an indicator of autophagy flux [70, 98]. In line with previous findings, we observed a clear reduction in LC3II levels in cells overexpressing VAPB compared to EGFP-transfected control and a significant accumulation of LC3II in Baf.A treatment compared to the control (Figure 7c). To further verify these results, we used a mouse fibroblast (NIH-3T3) cell line stably expressing EGFP-LC3 (see Materials and Methods), that was characterized by a baseline autophagic activity[71] showing both large and small LC3 punctae (Figure 7e, enlarged panel). Upon VAPB overexpression, we noted a decrease in the number and size of these LC3 punctae, indicating clearance of LC3 vesicles through autophagy induction (Figure 7e, enlarged panel and quantification)[70, 98]. Furthermore, increased levels of autophagosome-lysosome fusion proteins, including STX17 and SNAP29 [42] were representative of autophagy activation upon VAPB overexpression (Figure 7f, g, quantification h).
Overexpression of Wt-VAPB facilitates clearance of ALS-associated mutant RBP aggregates via autophagy.
Autophagy is the primary pathway for degrading misfolded proteins, and VAPB plays a crucial role in this process[109, 115]. Therefore, we hypothesized that VAPB could facilitate the degradation of pathogenic aggregates via activation of the autophagy pathway. To investigate this hypothesis, we utilized VAPB-induced autophagy to monitor the clearance of different types of ALS-associated toxic aggregates. We used mutant EGFP-P525L as well as Wt- FUS stable cell lines [75], in which P525L mutant FUS forms cytoplasmic aggregates (Figure 8a), which are also SDS insoluble (Figure 8a, lower panel). We expressed HA-VAPB or a control plasmid in these cell lines to assess autophagy flux and monitor FUS aggregation with or without VAPB. Western blot analysis of the cellular lysates confirmed the induction of autophagy by VAPB, evidenced by reduced levels of LAMP1 and LC3II (Figure 8b, d: quantification). Consistent with activated autophagy, VAPB expression significantly reduced the SDS-insoluble aggregates of FUS (Figure 8c, g: quantification). Using a similar approach on cell line overexpressing mutant (N-terminal deletion) - TDP43 (delta TDP-43) which forms cytoplasmic aggregate (Figure 8e), that can be biochemically resolved by FTA as SDS-resistant aggregates (Figure 8f). Overexpression of Wt-VAPB (HA-VAPB) reduced such TDP-43 aggregates (Figure 8f, quantification g).
Encouraged by these findings, we performed a similar set of experiments with the overexpression of Wt-VAPB and other FUS-ALS mutants and C9orf72-associated dipeptide repeat (DPR) aggregates causing mutants. C9orf72-associated dipeptide repeat (DPR) aggregates causing mutants also forms cytoplasmic and nuclear aggregates, that can be detected by FTA as well (Figure 8h). Consistent with earlier results, we observed a significant reduction in SDS-insoluble aggregation levels of these mutant proteins (Figure 8i, quantification). These results confirm the role of VAPB in regulating autophagy and managing the neuronal PQC.
{kind=link}
{kind=link}
{kind=link}