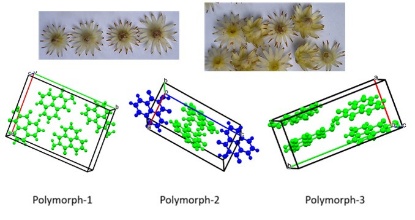
Structural comparisons among the polymorphs:
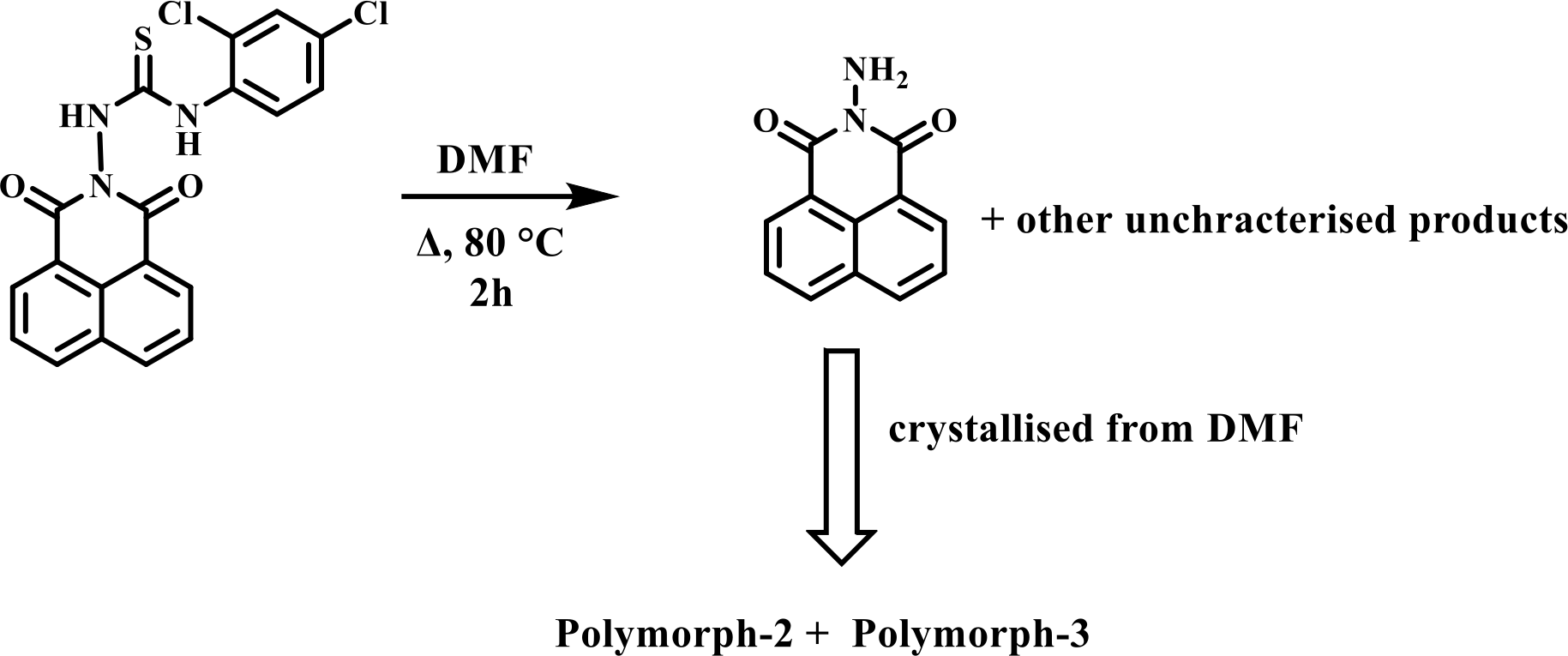
The crystals of the concomitant polymorphs of N-amino-1,8-naphthalimide were obtained serendipitously from the composition of thiourea derivative 1-(2,4-dichlorophenyl)-3-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)thiourea (DTU) at 80°C as illustrated in Scheme 1. The
powder pattern of the bulk crystals of the has all the principal peaks matching the theoretically generated pattern from the crystallographic information files of the two polymorphs as given in Fig. 5S. Our attempt to get only one form from the mixture was not successful. The crystals of the Polymorph-3 were picked up randomly and manually for structure determination, the crystals belonged to the orthorhombic Pna21 space group. The unit cell volumes of the three polymorphs of the N-amino 1,8-naphthalimide were similar. The unit cell parameters and crystal densities of are compared in Table 1. It may be mentioned that one of the reported polymorphs of N-amino-1,8-naphthalimide, designated here as Polymorph-2, had symmetry-independent molecules within the unit cell. In contrast, the other reported one, designated as Plymorph-1, had all symmetry-related molecules in the unit cell. The third form also contains all symmetry-equivalent molecules in the unit cell. The hydrogen-bonded N-amino-1,8-naphthalimide molecules of this polymorph are arranged as linear chains involving both the NH-NC = O faces of the pairs. The chain has
Table 1
Comparative unit-cell data of the three polymorphs
Polymorph | Polymorph-1 | Polymorph-2 | Polymorph-3 |
|---|
Space group | Orthorhombic Pna21 | Triclinic P -1 | Orthorhombic Pna21 |
Length of cell axis (Å) | a = 13.296(2), b = 18.408 (4), c = 3.781 (11) | a = 7.307(6), b = 9.364 (7), c = 14.655 (10) | a = 7.204(18), b = 16.373 (4), c = 7.996 (19) |
Angles (°) | α = β = γ = 90.00 | α = 81.676(3), β = 80.506(3), γ = 69.460(3) | α = β = γ = 90.00 |
Volume (Å3) | 925.5 (4) | 922.15 (12) | 943.1(4) |
Density (gcm− 3) | 1.518 | 1.523 | 1.495 |
hydrogen-bonded synthons with R22(10) graph-set notation as shown in Fig. 2a. The synthons have N2-H…O1{dD…A, 3.0579(8)Å; <DHA, 154°) and N2-H…O2 {dD…A, 3.0671(8)Å, <DHA, 159°} hydrogen bonds. The Polymorph-2 and Polymorph-3 have similarity in assembling, as both have chain-like structure; the difference arises from the two alternative pairs of symmetry independent molecules in the Polymorph-2, each associated as pairs with two independent R22(10) synthons (Fig. 1b), held to one another with distinguishable hydrogen bond parameters. The naphthalimide rings of independent neighboring chains of the Polymorph-3 are in parallel stacks. Considering the amino-group as the head and the naphthalene ring part as the tail, such stacking dipoles are aligned oppositely, showing a head-to-tail arrangement with a centroid-to-centroid distance of 3.649 Å. This distance is suggestive of weak interactions among the rings [44]. The Polymorph-1 had head-to-head arrangements, and Polymorph-2 had head-to-tail stackings in pairs formed among the neighboring naphthalimide rings. In the literature, there are examples of polymorphs with eclipsing 1,8-naphthalimide rings with dipoles aligned oppositely, as well as one having dipoles of rings oriented in the same direction [45]. On the other hand, different positional isomeric naphthalimide-based compounds having flexible tethers generate large variations in the stacks between the naphthalimide rings [46–47], and many polymorphs of naphthalimide derivatives with different orientations prepared by changing the crystallization process have wide variations in colors [48–49]. In the present example, the conformational adjustment can occur through the rotation of the N-N bond of the N-NH2 unit, changing the orientations of the hydrogen bond donor N-H bonds. In fact, this was a reason, together with the orientations of the rings in proximity, have caused the distinctions in the scheme of hydrogen bonds in packings of the three polymorphs.
Hirshfeld surfaces, total energies and DFT optimized energies:
The Hirshfeld surface analyses [50] of each polymorph were performed to distinguish the type of contacts in each case. The percentage of different interactions within a distance of 3.8 Å of the surface. The atoms in the vicinity of the surface are shown in Fig. 3. Different
percentages of contacts are listed in the supporting table and the pie-diagrams as for comparison are shown in Fig. 4a-c. They show that major contributions in the polymorphs were from contacts of hydrogen atoms and are comparable. The Allin-O in the three polymorphs was in order 2 > 1 > 3, and the Allin-C was 3 > 1 > 2. The Polymorph-1 and Polymorph-3 had identical nitrogen contacts with atoms inside the surface, whereas they had 0.8% less Allin-N contacts. This was due to differences in the orientation of the molecules of the symmetry-independent molecules within the lattice. Though the Polymorphs-1 and Polymorph-2 had differences in Allin-O and Allin-N, the total weight of these two contacts was higher in Polymorph-1 than the Polymorph-3, showing that the former had more amounts of moderate hydrogen bonds.
The dispersive and repulsive as well as total energies of the polymorphs by using Crystal Explorer with the B3LYP functional at the 6-31G(d,p) level. The respective dispersive energies are Polymorphs-1 to 3 were − 66.0 kJ/mol, -11.0 kJ/mol, and − 70.3 kJ/mol, whereas the respective repulsive energies were 30.8 kJ/mol, 21.2 kJ/mol, and 38.8 kJ/mol. Whereas the respective total energies − 48.7 kJ/mol, -25.6 kJ/mol, and − 51.2 kJ/mol. This showed that the stability of the polymorphs was of the order Polymorph-3 > Polymorph-2 > Polymorph-1.
The energy of each molecule in the conformation as observed in each polymorph was calculated by DFT using the B3LYP functional at the 6–31 + + G(d,p) level. It was found that each conformer has different energies, and the energies of the two symmetry-independent molecules differed by 5.2 kJ/mol from one another, as illustrated in Fig. 5a.. On the other hand electronic energy of the Polymorph 2 > 3 > 1. But the energy difference between the Polymorph-2 and Polymorph-3 was very small. The energy of each polymorph was also optimized in the gas phase by DFT calculation with B3LYP functional at 6–31 + + G(d,p) level. It was found that the optimized energies of Polymorph-1 and Polymorph-3 were the same; hence, the hydrogen-bonded dimers of Polymorphs-2 and Polymorph-3 were independently determined. This has revealed that polymorph-3 is 35.14 kJ/mol lower than the energy of Polymorph-1, whereas polymorph-2 is stable by 1.21 kJ/mol than Polymorph-3, as shown in Fig. 5, suggesting stability 2 > 3 > 1. These provided the stability gain by the synthons. A recent theoretical study has revealed that a high value of Z׳ (number of symmetry-independent molecules in unit cell) is not reflective of the stability of polymorphs [51]. Our observations suggest that the replica units of the assemblies, as well as the electronic energy of individual molecules, provide the trend of electronic stability of the discrete molecules in different conformations. The optimized energies of assembled structures within assemblies reflect to distinction of their relative gain or loss of stability by forming different synthons. But the total energy gain from dispersive and attractive forces provides insight into the overall stability of the polymorphs. In this case, Polymorph 3 has the lowest energy based on attractive and dispersive energies calculated by CrystalExplorer-17.5 version, and has the highest stability as reflected in the melting temperature. On the other hand, the density of polymorph-3 is lower but it has higher stability.
Differential scanning calorimetry and hot-stage microscopic study
The differential scanning calorimetry (DSC) of the concomitant polymorph was recorded, and it was compared with the DSC of the earlier reported ones. It showed that the Polymorph underwent melting of Polymorph-2 in the mixture at 248°C. Upon further heating, Polymorph-3 melted at 274°C. The first heating and cooling cycle of DSC is shown by the black trace in Fig. 6a. The corroboration of the results with the hot-stage microscopic study showed the melted phase, as shown in the (c) of Fig. 6. Upon cooling after heating up to 300°C, it showed that solidification of the melt at 262°C took place, which, on further cooling, reconverted to an amorphous phase at 221°C corresponding to polymorph-2. The second cycle of heating shown as the red trace of Fig. 6a showed the reversibility of the process, but the solidification at the second cycle took place at 259°C, which was 3°C lower than the first cycle. The images of the cooling samples at two temperatures are shown in Figs. 6b-e. This could be due to defects created within the structures after the first heating cycle, which had affected the colligative properties, affecting the solidification from the melt by forming another form of polymorph. The thermal properties are different from the other two polymorphs. Polymorph-1 melted at 249°C, whereas Polymorph-2 melted at 256°C. While cooling, the Polymorph-1 recrystallized at 233°C, whereas the
Polymorph-2 recrystallized at 253°C [42]. The thermogravimetric analysis shown in Fig. 7S has no weight loss on heating the concomitant polymorphs till 300°C (below this temperature, DSC plots were recorded), but there was a sharp loss of weight at 325°C, where the sample evaporated.
Structural studies on 1-(2,4-dichlorophenyl)-3-(1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)thiourea
We analyzed the structure of DTU from which the Polymorphs were formed, by determing the structure by X-ray single-crystal diffraction. The self-assembled structure of DTU is shown in Fig. 7a. The compound comprises a thiourea part linked to an N-amino 1,8-naphthalimide portion. The packing pattern shows that naphthalimide rings are not stacked but are in a slanted position, with a bisecting angle between the two planes containing such rings was 27.04°, as in (i) of Fig. 7a. Whereas the rings had occupied positions in an intermittent orientation, neither head to tail, nor head to head, as in (ii) of Fig. 7b. Hence, formation of the concomitant polymorph (oppositely having dipoles of the napthalimides) from this assembly will require flipping of one of the rings to reorganize to be parallel with the other and a rotation to make a head-tail orientation as suggested in Fig. 7b. In fact, the same argument would also suggest providing the other
polymorphs from such a decomposition. But the Polymorph-2 and Polymorph-3 had the highest comparable energies. Themogravimetry in Fig. 3S has shown that the compound DTU was stable up to 205°C; hence, polymorphs were formed as a result of a chemical decomposition caused by small amounts of water in the solvent or air that led to the formation of N-amino 1,8-napthalimide from DTU at 80°C. Furthermore, there is a literature example on cyclohexyl-derived thiocarbazide undergoing hydrolytic cleavage to produce a rearranged product [52]. It was also shown that thiocarbazide-based compounds result in different polymorphic solvates by changing crystallization conditions from room temperature to higher temperatures [43]. These observations have suggested that the specific polymorph crystallization was an outcome of the interplay of weak supramolecular interactions during the course of decomposition.
{kind=link}
{kind=link}