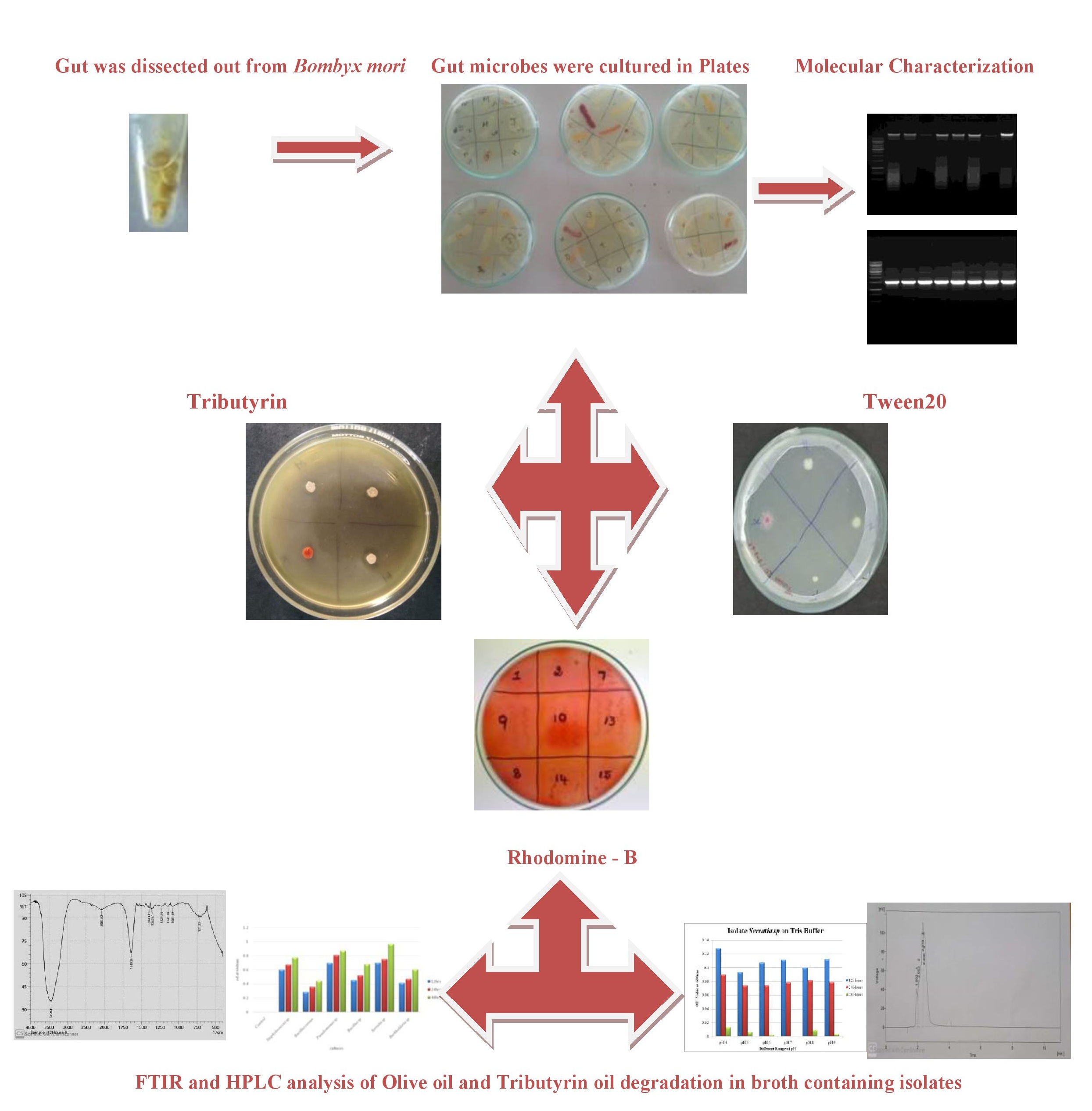
Sample Collection
Fifth instar silkworm larvae (Bombyx mori) were collected from the Central Silk Board [CSB], Extension Centre, Vaniyampatti, Srivilliputtur, Virudhunagar District, Tamil Nadu. Freshly collected fifth instar larvae were fed sterile young tender mulberry leaves. Silkworms were surface-sterilized with 75% ethanol for 1 min and rinsed three times with distilled water before dissection. The dissection was performed over a sterile wax-moulded plate using a dissection kit, such as a sterile needle, forceps, and scissors. The dissection of silkworm was performed to collect the midgut aseptically and store it in phosphate buffer saline (PBS) with a pH range between 7.4 and 8, as the midgut of Bombyx mori was in a similar range. PBS was used to maintain the microflora in the midgut of silkworms, and the collected midgut were stored in a freezer for further processing.
Isolation of the Migut Microflora
The stored midgut was thawed and gently vortexed. Then, the supernatant or liquid presented above the midgut is transferred. Then, using the supernatant, serial dilutions were prepared (from 10− 1 to10− 7) using PBS (Phosphate Buffer Saline). Using the spread plate, patch plate, and streak plate techniques, dominant colonies were isolated and used for further analysis of lipase screening and oil degradation
Screening of Lipase and Lipolytic Microorganisms
The isolated dominant colonies were then screened for lipolytic activity. Thus, the primary screening method involved plating dominant isolates on media enriched with lipid substrate. Thus, a sensitive plate assay technique was followed. Therefore, three different lipid substrate-supplemented media were used: Tween 20 agar medium and Tributyrin agar medium. The compositions of the media whereas follows: (i) Tween 20 agar medium: nutrient broth, 1.3 g/100 mL; Tween 20- 1-2% and agar- 2g/100mL; (ii) trichlorid agar medium (Himedia TBA medium were used, where Tributyrin oil was supplemented at the range of 1–2%).
Growth Curve Assay of Lipase and Oil using Bacteria
The growth curve assay was performed for the dominant and primarily screened lipolytic colonies, where the colonies were inoculated into two different Erlenmeyer flasks containing inoculum medium consisting of 0.5% beef extract, 0.5% peptone, 0.5% sucrose, 0.3% NaCl, 0.2% K2HPO4 and incubated at 30oC for 24 h on a rotary shaker at 200 rpm. After 24 h of incubation, the two-inoculum media were transferred into two other Erlenmeyer flasks containing production media consisting of 2.0% peptone, 0.5% sucrose, 0.1% (NH4)2SO4, 0.1% MgSO4, 0.2% K2HPO4, 1.0% olive oil and 2.0% peptone, 0.5% sucrose, 0.1% (NH4)2SO4, 0.1% MgSO4, 0.2% K2HPO4, 1.0% tributyrin oil; were incubated at 30oC on a rotary shaker at 200 rpm. The samples were collected after 12, 24, and 48 h, and the cell density was measured against the cell-free control by optical density at 600 nm using UV-Visible spectrophotometry.
Effect of pH
A single colony of gut isolates was inoculated in two different buffers, namely 100 mM phosphate buffer (Na2HPO4, NaH2PO4, NaCl & Distilled water) and 100 mM Tris HCl buffer (Tris HCl and distilled water), at different pH levels, such as pH 4,5,6,7,8,9 to understand the growth of lipolytic colonies under adverse conditions. Therefore, its efficiency in degrading lipid substrates, such as oil, can be studied.
Lipolytic activity analysis by FTIR spectroscopy
Fourier transform infrared spectroscopy (FTIR) is a technique used to obtain the absorption or emission spectrum of a solid, liquid, or gas. In FTIR analysis, the isolated bacterial colonies were studied for the lipolytic activity on olive oil and tributyrin oil at different time periods of incubation, such as 12 h, 24 h, and 48 h.
HPLC Analysis
The isolated colonies were incubated in olive oil production medium. After 24 h of incubation, the production medium with the culture was centrifuged at 10,000 rpm for 15–20 min to settle down the cells at the bottom. After centrifugation, the supernatant was transferred, dissolved in an equal volume of methanol, and centrifuged as necessary. The solvent was then allowed to undergo high-performance liquid chromatography (HPLC).
Molecular Identification of Potent Lipolytic Isolates
Genomic DNA preparation
The sonicated gut suspension was subjected to serial plating. Microbial genomic DNA was extracted following the protocol described by Ausubel et al. 1994. The gut susppension was subjected to chemical and enzymatic rupture by adding 90µl of 10% SDS and 90µl lysosyme (20 mg/ml) (gently mixed). The tubes were incubated at 37°C for 1.5 h each tube was mixed with 150µl of 5M NaCl before the addition of 100µl of 5M NaCl containing 10% cetyltrimethyl-ammonium bromide. The samples were thoroughly mixed and incubated at 65°C for 3 min. DNA was extracted using phenol: chloroform: isoamyl alcohol (25:24:1[vol/vol/vol]). DNA was precipitated using 70% ethanol and recovered by centrifugation (Broderick et al. 2004). Pellets were resuspended in 20µl of TE buffer.
Template preparation for microbial fingerprinting analysis
The bacterial colonies picked up from the subcultured plates were inoculated into 1 ml of TSB broth and shaken for 3 h at 200 rpm and 37°C. After turbidity development, the tubes were centrifuged at 10,000 rpm for 10 min. The pellet obtained was subjected to treatment with 50 µl of colony lysis buffer (CLB) (1% Triton X-100, 20mM Tris HCl pH 8.0, 2mM EDTA pH 8.0). The mix was vortexed for a few seconds and heated at 65°C for 15mins, then 5 µl from each tube was transferred to fresh tubes as the template for microbial fingerprinting analysis.
BOX PCR amplification
BOX PCR was performed to identify the microbial footprints of BOX elements in specific strains. The reaction mixture contained 5 µl of 10X PCR buffer, 10 mM dNTP mixture (2.5mM each), 1.0 µM primer, 1.5 unit Taq DNA polymerase, and 50 ng template DNA. Amplification was performed with a final volume of 50 µl using a thermal cycler (ABI). The template was subjected to initial denaturation at 94°C for 4 min, final denaturation at 94°C for 1 min, primer annealing at 60°C for 2 min, and extension of the reaction primer was extended at 74°C for 2 min. The steps were repeated for 40 cycles, and the final extension was performed at 74°C for 5 min. PCR-amplified fragments were electrophoresed on 2% agarose gel and examined under UV light after staining with ethidium bromide.
ERIC PCR amplification
Enterobacterial repetitive intergenic consensus sequence (ERIC) primer amplification was used to identify significant diversity and compare the fingerprints of 16S rRNA genes in the gut community. A 25 µl reaction mixture comprises of 1.0 µM upstream primer Eric F and downstream primer Eric R, 10 PCR buffer, 2.0 µl 10 mM dNTP mixture (2.5 mM each), 1.5-unit thermostable Taq DNA polymerase, and 50 ng of template DNA. The template was subjected to initial denaturation at 94°C for 4 min and final denaturation at 94°C for 1 min, followed by primer annealing at 60°C for 2 min, and the reaction primer was extended at 74°C for 2 min. The steps were repeated for 40 cycles, and the final extension was performed at 74°C for 5 min. The amplified products were electrophoresed on 2% agarose gel and examined under UV light after staining with ethidium bromide.
16S rRNA amplification
To amplify 16S rRNA, the universal eubacterial primers 27F and 1492R were used. The reaction was performed under the following conditions using a 27F forward primer and 1492R reserve. The final volume of the mix was 25 µl contains 5 µl of 10X reaction buffer, 2.5 µl of 10 mM dNTP mixture, 1 µl of 10 µM forward and reverse primers, 2.5 units Taq polymerase, and 50mg template DNA. The initial denaturation at 94ºC for 3 min was cycled for 35 reactions with denaturing the template for 30 sec at 94ºC, annealing at 55ºC for 1.5minute, the reaction was extended for 2.5 min at 72ºC and the reaction was finally extended for 10 min at 72ºC.
{kind=link}