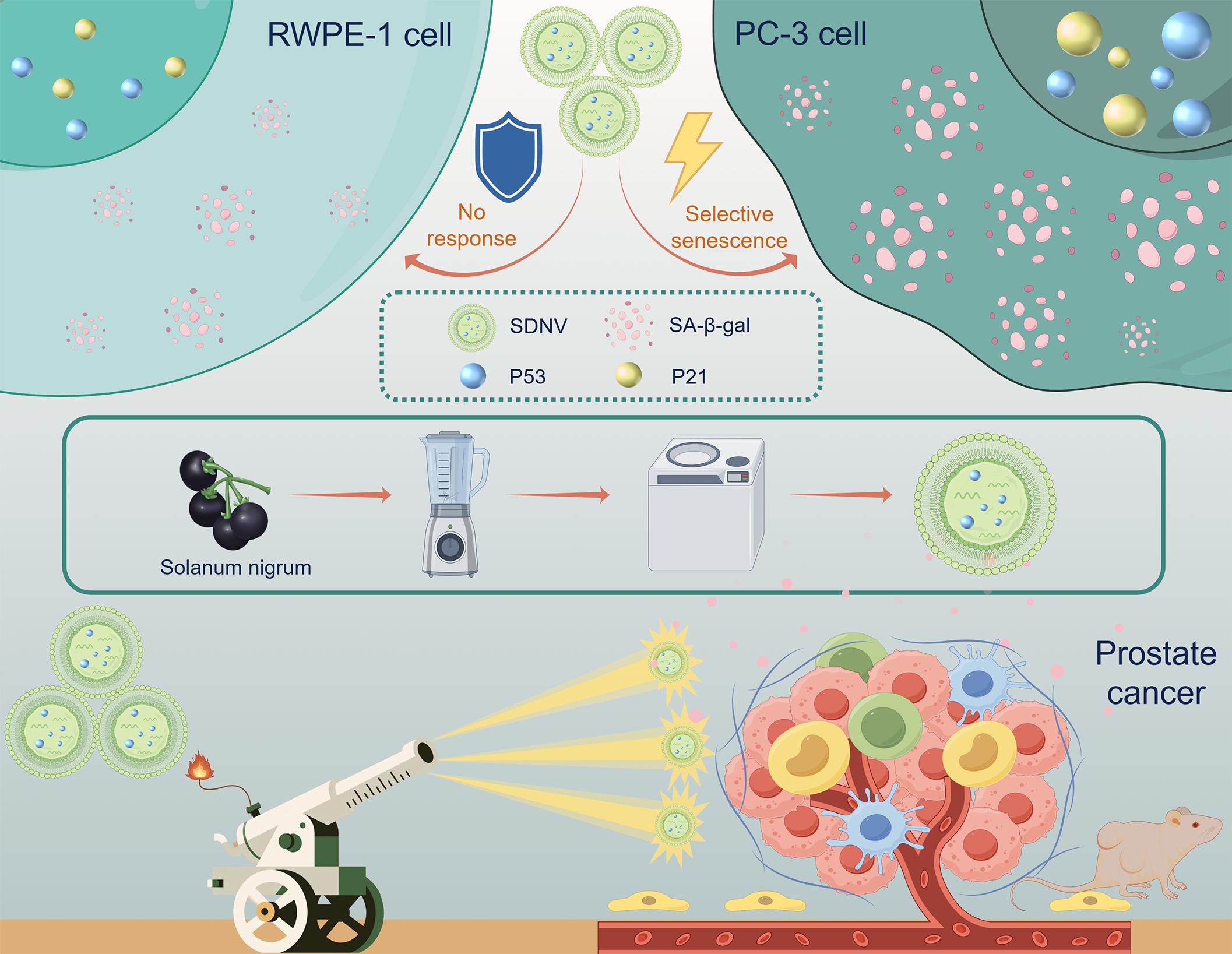
2.1 Isolating, purifying, and characterizing SDNVs
The dried Solanum nigrum fruits were homogenized using a blender with an appropriate volume of double-distilled water for 3 minutes to obtain the crude extract. The supernatant was collected through differential centrifugation at 300 ×g for 10 minutes, 2,000 ×g for 20 minutes, and 10,000 ×g for 30 minutes sequentially, with the supernatant retained after each step. The resulting supernatant was subjected to ultracentrifugation at 135,000 ×g for 70 minutes. The pellet was resuspended in 20 mmol/L Tris-HCl buffer to yield a yellow suspension enriched in SDNVs. The crude SDNVs were further purified using a discontinuous sucrose gradient. Sucrose solutions of 15%, 30%, 45%, and 60% (w/v) in double-distilled water were prepared. The sample was layered at the bottom of the centrifuge tube, followed by careful overlaying of each sucrose layer in ascending order. After centrifugation at 150,000 ×g for 2 hours, a visible SDNV-enriched band was collected from the 15–30% interface and subjected to a second round of ultracentrifugation at 150,000 ×g for 2 hours. The final pellet was resuspended in Tris-HCl buffer and filtered through a 0.22 µm sterile membrane to obtain a clarified, sterile SDNV suspension.For morphological assessment, SDNVs were negatively stained and examined by transmission electron microscopy (TEM) using a Tecnai G2 Spirit Twin microscope equipped with a Gatan 832.10W camera. Particle size distribution and concentration of SDNVs were determined using nanoparticle tracking analysis (NTA) on a NanoSight NS300 instrument. Total protein content of SDNVs was determined using a BCA assay kit (Biosharp, BL521A) according to the manufacturer’s instructions. Protein identity and purity were further evaluated using Coomassie brilliant blue staining. Total RNA content was quantified using an Agilent 2100 Bioanalyzer.
2.2 Metabolomics analysis
Metabolomic profiling of SDNVs was performed using ultra-high-performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS). Briefly, SDNV samples, water decoction of Solanum nigrum (SWD), and selected reference standards (Solasonine) were treated with methanol for protein precipitation, followed by centrifugation to collect the supernatants. These were subsequently analyzed by UHPLC-MS/MS. All analyses were carried out on a Vanquish UHPLC system coupled to an Orbitrap Q Exactive™ HF or HF-X mass spectrometer (Thermo Fisher Scientific, Germany) at Novogene Co., Ltd. (Beijing, China). Raw data were processed using Compound Discoverer software (version 3.3, Thermo Fisher Scientific), including peak extraction, retention time alignment, compound identification, and relative quantification.
2.3 Labeling of SDNVs
For fluorescent labeling, SDNVs (equivalent to 100 mg total protein in 1 mL PBS) were incubated with 1 mL of DiR (Macklin, D909616), DiI (Macklin, D82309), or DiO dye (Zeta Life, DiO-10), each at a concentration of 1 mM in DMSO, at 37°C for 30 minutes. After labeling, excess unbound dye was removed by ultracentrifugation at 135,000 ×g for 70 minutes. The labeled SDNV pellet was washed and resuspended in sterile PBS.
2.4 Cell culture
The human prostate cancer cell line PC-3 and normal human prostate epithelial cell line RWPE-1 were obtained from ICELL (Shanghai, China). PC-3 cells were cultured in RPMI-1640 medium (Gibco, 11875-093) supplemented with 10% fetal bovine serum (FBS) (Gibco, 10099141) and 1% penicillin-streptomycin (Gibco, 15140122). Cells were maintained at 37°C in a humidified incubator with 5% CO₂. RWPE-1 cells were cultured in Keratinocyte Serum-Free Medium (K-SFM) (Gibco, 17005042), supplemented with 0.05 mg/mL bovine pituitary extract (BPE) and 5 ng/mL recombinant human epidermal growth factor (EGF), according to the manufacturer's protocol. Cells were maintained under the same incubation conditions. Cells were routinely subcultured at 70–80% confluence and used for experiments within 10 passages.
2.5 uptake of SDNVs by PC-3 cells
To evaluate cellular uptake, PC-3 cells were seeded in 6-well plates or glass-bottom confocal dishes and cultured until 70–80% confluence. For confocal microscopy, cells were incubated with DiI-labeled SDNVs (1 × 10⁹ particles/mL) for 6 hours, then washed with PBS, fixed with 4% paraformaldehyde, and stained with DAPI. Internalization and intracellular localization were visualized using laser scanning confocal microscopy (LSM800, Zeiss, Germany). To assess uptake efficiency, PC-3 cells were incubated with DiO-labeled SDNVs for 0, 1, 3, 6, 12, and 24 hours, respectively. After incubation, cells were harvested, washed, and analyzed by flow cytometry (BD FACSCanto II) using the FITC channel. All experiments were performed in triplicate.
2.6 Cell viability assay
PC-3 cells were seeded in 96-well plates at a density of 5 × 10³ cells/well and allowed to adhere overnight. Cells were then treated with various concentrations of SDNVs (0, 12, 18, 24, 30, 36, 48 µg/mL) for 24 hours. After treatment, 10 µL of Cell Counting Kit-8 (CCK-8, Dojindo, CK04) solution was added to each well and incubated for 3 hours at 37°C. Absorbance was measured at 450 nm using a microplate reader (BioTek, Synergy H1). Cell viability was calculated relative to the untreated control group. All assays were conducted in triplicate..
2.7 Cell proliferation assay
To further evaluate the antiproliferative effects of SDNVs on PC-3 cells, an EdU incorporation assay was performed using the Cell-Light™ EdU Apollo 488 kit (Beyotime, C0078S). PC-3 cells were seeded into 6-well plates containing sterile coverslips and treated with SDNVs (36 µg/mL) for 24 hours. EdU was diluted 1:500 from a 10 mM stock solution into complete culture medium to prepare a 2× working solution (20 µM), which was added in equal volume to the wells to yield a final concentration of 10 µM EdU. Cells were incubated at 37°C for 2 hours. After incubation, cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature, then washed three times with PBS containing 3% bovine serum albumin (BSA). Permeabilization was performed using 0.3% Triton X-100 in PBS for 15 minutes, followed by another PBS/BSA wash. The “click” reaction mixture was freshly prepared by combining the following components per 1 mL: 860 µL Click Reaction Buffer, 40 µL CuSO₄, 2 µL Azide 488, and 100 µL Click Additive Solution. A total of 100 µL of the reaction cocktail was added to each well and incubated in the dark at room temperature for 30 minutes. After three washes, nuclear staining was performed using Hoechst 33342 (1:1000 dilution in PBS) for 10 minutes. EdU-positive cells (green fluorescence) and total nuclei (blue fluorescence) were visualized using a fluorescence microscope. The proliferation rate was calculated as the ratio of EdU-positive cells to total nuclei. All experiments were performed in triplicate.
2.8 Cell migration assay
The effect of SDNVs on PC-3 cell migration was evaluated using a Transwell assay. PC-3 cells were harvested using 0.25% trypsin, washed, and resuspended in serum-free medium. A total of 1.5 × 10⁴ cells in 100 µL serum-free medium were seeded into the upper chamber of a Transwell insert (8.0 µm pore size; Corning, USA). The lower chamber was filled with 400 µL of complete medium containing 10% fetal bovine serum as a chemoattractant. The plates were incubated at 37°C in a humidified incubator with 5% CO₂ for 12 hours. After incubation, non-migrated cells on the upper surface of the membrane were gently removed with a cotton swab. The inserts were rinsed twice with PBS, and the migrated cells on the lower surface were fixed with 4% paraformaldehyde for 10 minutes. After washing with distilled water, cells were stained with 1% crystal violet solution (filtered through a 0.45 µm filter) for 10 minutes, followed by PBS washes to remove excess stain. Migrated cells were imaged and quantified under an inverted microscope (Olympus CKX53). Five random fields per membrane were captured and counted to evaluate the average number of migrated cells. All experiments were conducted in triplicate.
2.9 Cell apoptosis assay
Apoptosis of PC-3 cells was assessed using an Annexin V-AbFluor™ 488/PI apoptosis detection kit (Abbkine, KTA0002) according to the manufacturer’s protocol. After treatment with SDNVs (36 µg/mL) for 24 hours, cells were harvested using trypsin without EDTA and centrifuged at 300 ×g for 5 minutes at 4°C. The cell pellet (1–2 × 10⁵ cells) was washed twice with pre-chilled PBS and resuspended in 1× binding buffer. Annexin V-AbFluor™ 488 and propidium iodide (PI) were added to the cell suspension and incubated for 15 minutes in the dark at room temperature. Samples were analyzed within 30 minutes using flow cytometry (BD FACSCanto II). The percentage of apoptotic cells (early + late) was quantified. All experiments were conducted in triplicate.
2.10 Nude mouse xenograft model:
Male BALB/c nude mice (8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained under specific pathogen-free (SPF) conditions with ad libitum access to food and water. After a one-week acclimatization period, each mouse was subcutaneously injected with 1 × 10⁷ PC-3-Red-Fluc prostate cancer cells suspended in 200 µL of PBS into the right flank. When the tumor volume reached approximately 20 mm³, mice were randomly divided into four groups (n = 6 per group): (1) Control group: gavaged with 100 µL PBS; (2) Low-dose SDNV group: gavaged with 100 µL of SDNV suspension (0.18 µg/µL); (3) Medium-dose SDNV group: gavaged with 100 µL of SDNV suspension (0.54 µg/µL); (4) High-dose SDNV group: gavaged with 100 µL of SDNV suspension (1.62 µg/µL). Oral administration was performed once daily for 28 consecutive days. At the end of the treatment period, mice were euthanized, and tumor tissues were excised, weighed, and processed for subsequent histological and molecular analyses. Tumor volume was measured every three days using a caliper and calculated with the formula: V = ½ × length × width².
2.11 Tumor histopathology and immunohistochemical analysis
Hematoxylin and eosin (H&E) staining was performed to evaluate tumor architecture, necrosis, and cellular morphology. Apoptotic cell death was assessed using the TUNEL assay. Cell proliferation was evaluated via immunohistochemical staining for Ki-67. After deparaffinization and rehydration, antigen retrieval was performed in citrate buffer (pH 6.0) at 95°C for 15 minutes. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide, followed by incubation with anti-Ki-67 primary antibody (Servicebio, GB111499; 1:500 dilution) overnight at 4°C. After incubation with HRP-conjugated secondary antibody, staining was developed using DAB substrate, and sections were counterstained with hematoxylin. Positive Ki-67 staining was visualized as brown nuclear staining and quantified as the percentage of positive nuclei among total nuclei in five random fields.
2.12 Biodistribution of Orally Administered SDNVs
The experimental approach employed for assessing the in vivo distribution of PDNVs builds upon prior research conducted by our team [12]. When tumors reached approximately 20 mm³, DiR-labeled SDNVs were administered by oral gavage once daily at a dose of 1 × 10¹¹ particles/kg for 7 consecutive days. Six hours after the final gavage, the animals were euthanized, and major organs—including the heart, liver, spleen, lungs, kidneys, and tumor—were harvested for ex vivo fluorescence imaging. Tissue fluorescence was visualized and quantified using a Tanon ABLX3 in vivo imaging system (Tanon, Shanghai, China). The intensity of DiR signal in different organs was used to evaluate the biodistribution pattern of orally administered SDNVs.Tissue
2.13 Biosafety evaluation of Orally Administered SDNVsAt the end of the treatment period, major organs including the heart, liver, spleen, lungs, and kidneys were harvested from each mouse, rinsed with PBS, and fixed in 4% paraformaldehyde for 24 hours. Fixed tissues were dehydrated, embedded in paraffin, and sectioned at 5 µm thickness. Hematoxylin and eosin (H&E) staining was performed following standard protocols. Histopathological changes were examined under a light microscope to evaluate potential tissue toxicity.To assess systemic biosafety, body weight of mice was recorded To To assess systemic biosafety, body weight of mice was recorded every 3 days during the 4-week treatment period. Blood samples were collected by cardiac puncture immediately after euthanasia. Serum was isolated by centrifugation at 1,500 ×g for 15 minutes and subjected to biochemical analysis using an automatic biochemical analyzer to measure liver and kidney function markers, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE).
2.14 Transcriptomic analysis of SDNV-treated cells
Total RNA was extracted using TRIzol reagent (Invitrogen, USA), and RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). RNA libraries were constructed using the NEBNext® Ultra™ Directional RNA Library Prep Kit (NEB, USA) and sequenced on an Illumina NovaSeq 6000 platform (150 bp paired-end reads). Raw reads were quality-filtered using fastp and mapped to the human reference genome (GRCh38) using HISAT2. Gene expression was quantified using featureCounts and normalized to fragments per kilobase per million mapped reads (FPKM). Differential expression analysis was conducted with the DESeq2 R package. Genes with |log₂(fold change)| ≥ 1 and adjusted p-value < 0.05 were considered significantly differentially expressed. KEGG pathway enrichment analysis was performed using clusterProfiler (v4.6.2). Gene Set Enrichment Analysis (GSEA) was conducted using the GSEA software (v4.3.2) with the MSigDB c2.cp.kegg.v7.5.1 gene set database.
2.15 β-Galactosidase Staining Assay
Senescence-associated β-galactosidase (SA-β-gal) staining was performed to evaluate the induction of cellular senescence in PC-3 cells following SDNV treatment. Cells were seeded into 6-well plates and treated with SDNVs (50 µg/mL) or PBS (control) for 24 hours. After treatment, the culture medium was removed, and cells were washed once with PBS. Cells were then fixed with 1 mL of β-galactosidase fixative solution (Beyotime, C0602) for 15 minutes at room temperature. Following fixation, cells were washed three times with PBS (3 minutes per wash), and 1 mL of freshly prepared staining working solution was added to each well. The staining solution was composed of 10 µL of solution A, 10 µL of solution B, 930 µL of solution C, and 50 µL of X-Gal stock solution (20 mg/mL). Plates were sealed with parafilm and incubated overnight at 37°C in a dry incubator (without CO₂). The following day, SA-β-gal-positive cells exhibiting blue-green cytoplasmic staining were observed under a light microscope. Senescent cells were quantified in five randomly selected fields per well. Experiments were performed in triplicate.
2.16 Quantitative real-time PCR
Total RNA was extracted from PC-3 cells using Total RNA Extraction Reagent (Vazyme, R401-01) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized using a reverse transcription premix kit (AG11728, Accurate Biology, China). Quantitative real-time PCR (qPCR) was performed using the SYBR Green Pro Taq HS Premixed qPCR Kit (AG11701, Accurate Biology, China) on a LightCycler 480 system (Roche, Switzerland). The expression levels of senescence-related genes including TP53, P21, and P16 were analyzed. ACTB was used as the internal reference. Relative gene expression was calculated using the 2⁻^ΔΔCt^ method. Each sample was analyzed in triplicate, and data were expressed as mean ± standard deviation (SD). The primer sequences are listed in Additional file 1: Table S1.
2.17 Western Blot Analysis
Total protein was extracted using RIPA lysis buffer (Beyotime, P0013B) supplemented with protease and phosphatase inhibitors (Beyotime, P1045). Protein concentration was determined using a BCA Protein Assay Kit (Biosharp, BL521A). Equal amounts of protein (20–30 µg) were separated by SDS-PAGE on 10% polyacrylamide gels and transferred onto PVDF membranes (Millipore, IPVH00010). Membranes were blocked with 5% non-fat milk in TBST at room temperature for 1 hour and then incubated overnight at 4°C with primary antibodies: anti-P53 (ZENBIO, 345567, 1:1000), anti-P21 (ZENBIO, 381102, 1:1000), and anti-GAPDH (Proteintech, 60004-1-Ig, 1:5000). After washing, membranes were incubated with HRP-conjugated secondary antibodies (EARTH, E030110-01, 1:10000; HUABIO, HA1001, 1:10000) for 1 hour at room temperature. Protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit (Biosharp, BL520A), and images were captured using a Tanon 5200 imaging system. The relative expression levels of target proteins were quantified by densitometric analysis using ImageJ software and normalized to GAPDH.
2.18 Statistical Analysis
All experimental data are expressed as the mean ± SD. Statistical analyses were performed using GraphPad Prism software (version 9.0, GraphPad Software, USA). Comparisons between two groups were conducted using unpaired two-tailed Student’s t-tests, while one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used for comparisons among three or more groups. A p-value < 0.05 was considered statistically significant. Significance levels in figures are indicated as follows: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and ns for non-significant differences (p > 0.05).
{kind=link}