Tissue samples
Eighty GC tissue samples and the corresponding adjacent normal tissues were obtained from the First Affiliated Hospital of Nanjing Medical University between April 2015 and April 2021. All patients were definitively diagnosed by at least two pathologists to have no history of preoperative chemoradiotherapy or other malignancies. The clinicopathological characteristics of the patients were obtained from medical records, and the survival status was ascertained through annual telephone interviews. After informed consent was obtained from each participant, our study was evaluated and approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University.
Cell culture
The cell lines used in this study, including HEK-293T, the normal human glandular epithelial cell line GES-1, and several common human GC cell lines, were provided by the Cell Center of Shanghai Institutes for Biological Sciences. HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM H-21 4.5 g/litre glucose) (Wisent, Canada), and AGS cells were cultured in Nutrient Mixture F-12K medium (Wisent, Canada). The other aforementioned cell lines were cultured in RPMI 1640 medium. Foetal bovine serum (FBS; 10%, Wisent, Canada) was added to the aforementioned media along with 1% penicillin-streptomycin. All cell lines were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Plasmid construction, siRNA transfection, and lentiviral infection
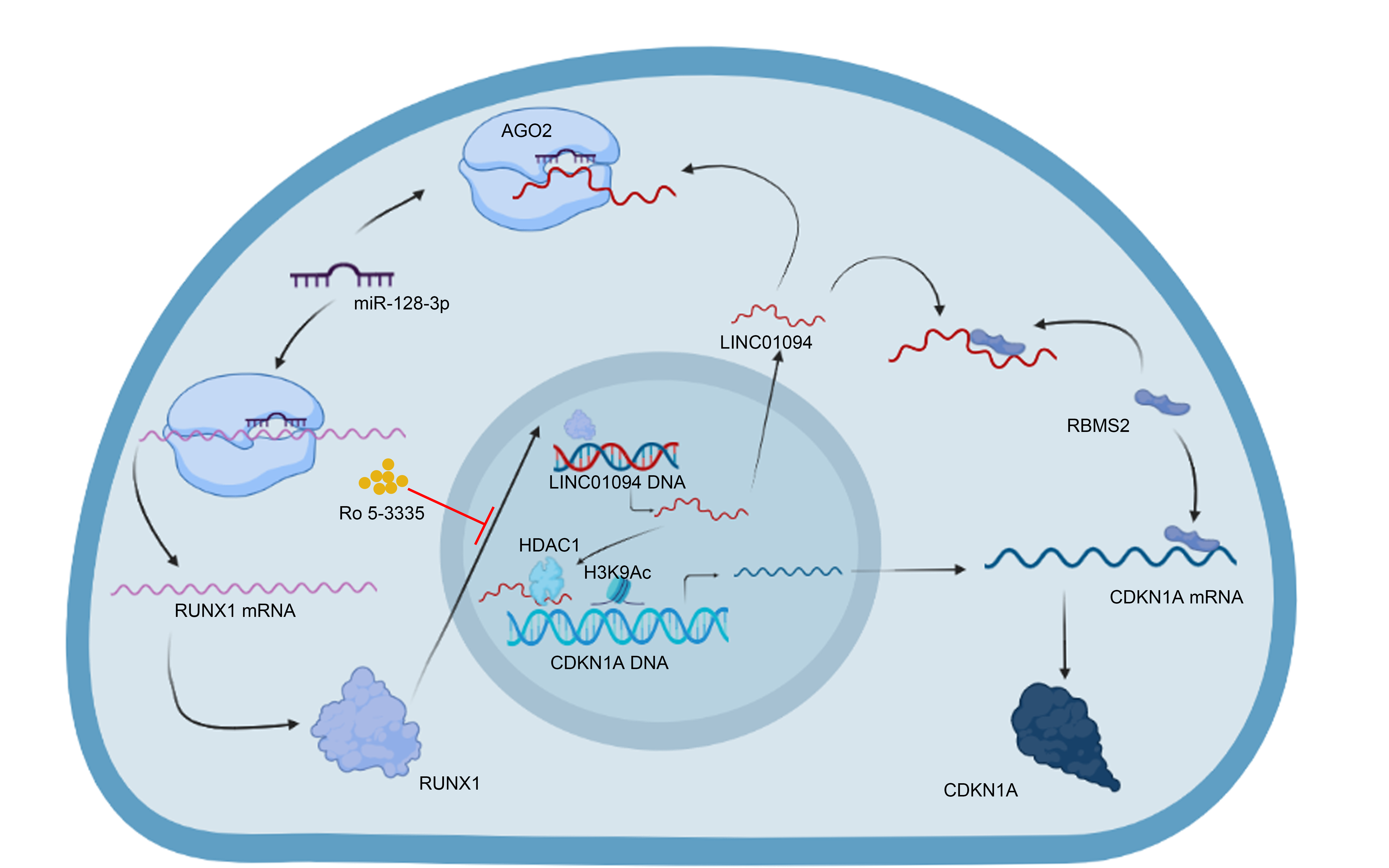
The human LINC01094 overexpression vector was constructed by inserting the corresponding cDNA sequence into the pcDNA3.1 vector (Obio, China), with the empty vector used as a control. Small interfering RNAs (siRNAs) against LINC01094 (GenePharma, China) were synthesized, with a scrambled siRNA used as a negative control. Lipofectamine 3000 (Invitrogen, USA) was used for transfection. The sequences of the LINC01094 cDNA and the siRNAs targeting LINC01094 and cyclin-dependent kinase inhibitor 1A (CDKN1A) were inserted into a lentiviral vector (GenePharma, China). Specific plasmids expressing and siRNAs targeting RNA binding motif single stranded interacting protein 2 (RBMS2), histone deacetylase 1 (HDAC1), CDKN1A, and RUNX family transcription factor 1 (RUNX1) were constructed as described above. Expression at different levels was evaluated by quantitative real-time reverse transcription polymerase chain reaction (qRT‒PCR) to validate the gene knockdown and overexpression efficiencies in the supplementary material (Fig. S1A-B and Fig. S3A-D). The related sequences were used in previous studies and are listed in the supplementary material [19–25].
RNA extraction and qRT-PCR
Total RNA was extracted from cells and tissues by TRIzol reagent (Invitrogen, USA). The New Poly (A) Tailing Kit (Thermo Fisher Scientific, USA) and the Prime Script RT Reagent Kit (Takara, Japan) were used for the reverse transcription of miRNA and lncRNA/mRNA, respectively. Amplification was performed with SYBR Green Master Mix (Roche, China). LncRNA and mRNA expression were normalized to GAPDH expression. Related procedures were performed three times, and the 2−ΔΔCT method was applied to evaluate expression levels. The primers used in this study are described in the supplementary material.
Nuclear-cytoplasmic fractionation
The PARIS™ kit (Thermo Fisher Scientific, USA) was used for isolation of the nuclear and cytoplasmic fractions according to the manufacturer’s steps. After lysing and centrifuging the cells, we obtained the supernatant as the cytoplasmic fraction. Subsequently, we washed the pelleted nuclei with a Cell Fraction Buffer and collected the nuclear fraction.
mRNA stability and protein stability analyzes
For mRNA stability analysis, we treated MKN45 cells with 5 µg/mL actinomycin D (ActD; Sigma-Aldrich, USA) and measured target gene expression at 2-hour intervals (to 12 h). Cycloheximide (CHX; 50 µg/mL; Sigma-Aldrich, USA) was used for protein stability analysis. The remaining procedures were similar to those described above.
Fluorescence in situ hybridization (FISH) assay
Fluorescently labelled LINC01094 and miR-128-3p probes were constructed, and 4’,6-diamidino-2-phenylindole (DAPI) was applied to stain nuclei. The probe signals in GC cells and tissues were detected with a FISH kit (RiboBio, China). Images were obtained with confocal microscopy (Carl Zeiss, Germany).
RNA pull-down assay
According to the instructions, we incubated a biotin-labeled LINC01094 probe (RiboBio, China) with streptavidin magnetic beads. After probe-coated beads were generated, we obtained MKN45 cell lysates, incubated them with the aforementioned beads overnight, and then washed the beads. Then, we performed silver staining with a silver staining kit (Thermo Fisher Scientific, USA). Furthermore, we performed mass spectrometry and western blotting to analyze the precipitated proteins.
RNA immunoprecipitation (RIP) assay
Following the protocol of the Magna RIP RNA Binding Protein Immunoprecipitation Kit (Millipore, USA), we incubated magnetic beads with specific antibodies at 25°C and mixed the GC cell lysates with the antibody-bead complexes at 4°C. Then, qRT-PCR was applied to quantify the eluted bound RNAs.
Chromatin immunoprecipitation (ChIP) assay
We purchased the Magna ChIP™ A/G Kit (Millipore, USA) for the ChIP assay. After fixing the cells with 1% formaldehyde and quenching the crosslinking reaction, we generated 200-1000-bp fragments of the crosslinked chromatin by sonication. The magnetic beads were incubated with related antibodies and cell lysates. Then, qRT-PCR was applied to analyze the eluted immunoprecipitated complexes.
Triplex capture assay
We incubated MKN45 cell nuclei with 2 µg biotinylated triplex-forming oligonucleotides (TFOs) and subjected them to UV (365 nm) irradiation and sonication. After centrifugation of the nuclei, the supernatant was separated and incubated with streptavidin-magnetic beads (Thermo Fisher Scientific, USA) at 4°C. Following DNA extraction and purification and qRT-PCR analysis, we washed the beads 5 times and then resuspended the eluted complexes. The primers used are described in the supplementary material.
Luciferase reporter assay
The corresponding fragments were synthesized and subcloned into the luciferase reporter plasmid (Realgene, China). Various gene overexpression plasmids and siRNAs were cotransfected with the luciferase reporter plasmid. Then, we collected the cells and applied the Dual-Luciferase Assay Kit (Promega, USA). Firefly and Renilla luciferase activities were measured and are presented as relative luciferase activity values (firefly luciferase/Renilla luciferase).
Western blot analysis
The western blot protocol was consistent with that used in our previous study, and the antibodies used are described in the supplementary material [26].
Immunohistochemical (IHC) and hematoxylin & eosin (HE) staining
For IHC staining, paraffin-embedded tissues were dewaxed, rehydrated, and subjected to antigen retrieval. Specific antibodies were incubated with the sections, and expression levels were evaluated with diaminobenzidine (DAB) the next day.
Paraffin-embedded mouse lung tissues were sliced into 5-µm sections and stained with HE for pathologic analysis.
Functional assays
Cell proliferation, migration, and invasion assays
To observe the effects on cell proliferation, migration, and invasion, Cell Counting Kit-8 (CCK-8), colony formation, wound healing, and transwell assays were performed as described in our previous study [26].
Flow cytometric analysis
The Annexin V-FITC/PI Apoptosis Detection Kit (Multisciences, China) and the Cell Cycle Analysis Kit (Multisciences, China) were applied to evaluate the apoptosis rate and the cell cycle, respectively, according to the corresponding protocols. The apoptosis rate was calculated as the sum of early apoptotic and late apoptotic cells. Cell cycle data were divided into G1, S as well as G2/M phases and analyzed by FlowJo (v10.8.1).
Animal experiments
Cell suspensions (0.1 ml, 1 × 106 stable cells) were subcutaneously injected into four-week-old male nude mice (BALB/c) to establish the xenograft model. We measured the tumor volume every 2 days and weighed the xenografts after sacrificing the mice 2 weeks after cell injection. For the lung metastasis model, we injected cells into the caudal vein of nude mice, and metastatic tumors were detected by HE staining. The care of animals involved in this study was in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals.
GC organoid model
Human GC organoids model were constructed as described previously [27]. We transfected the LINC01094 shRNA and its negative control into the organoids to facilitate our investigation of the role of LINC01094 in GC progression. The growth of GC organoid was observed daily by microscope.
Bioinformatic and statistical analyzes
All DNA, RNA, and protein sequences used in this study can be searched in the National Center for Biotechnology Information (NCBI) database. Other public databases and the corresponding websites are listed in the supplementary material. SPSS (version 26.0) and GraphPad Prism (version 8.01) were used for statistical analysis. Student's t test, the Wilcoxon test, and the χ2 test were applied to analyze parametric variables, and the Kaplan‒Meier method was employed to analyze overall survival (OS). Quantitative data are presented as the means ± SDs. Each experiment was repeated independently in triplicate, and p < 0.05 was considered to indicate statistical significance.
{kind=link}