3.1. Taxonomic Affiliation and Phylogeny of the Metagenome-Assembled Genomes
Obtaining metagenomic DNA for further genome reconstruction from hydrocarbon-contaminated samples is usually complex due to the high contaminant concentration and low microbial biomass present. In order to recover as many MAGs as possible, a metagenomic pipeline with four binning tools followed by a dereplication tool was used. The assembly and genome recovery of the six paired-end metagenome datasets yielded 112 non-redundant MAGs. The quality check classified nine MAGs as medium-quality (completeness > 50% and contamination < 10%), while 11 MAGs were classified as high-quality (completeness > 90% and contamination < 5%), according to the MIMAG standard(Bowers et al. 2017) (Fig. S2). Low-quality MAGs were filtered and not used for further analyses. Nineteen of twenty MAGs selected belonged to the Bacteria domain, while only one was affiliated to the Archaea domain. The most representative bacterial phyla were Bacteroidota (n = 9) and Patescibacteria (n = 4), followed by Firmicutes (n = 2), Proteobacteria (n = 2), Desulfobacterota (n = 1), Thermoproteota (n = 1) and Actinobacteriota (n = 1) (Table S2). Several studies have showed that Proteobacteria, Firmicutes and Bacteroidota phyla are predominant and play key roles in petroleum biodegradation in impacted areas(Hidalgo et al. 2019; Shahi et al. 2016). In addition, members of the recently proposed phylum Patescibacteria(Parks et al. 2018), characterized by small cell and genome sizes(Tian et al. 2020), are often found in polluted aquifers and environments(Antunes et al. 2021; Hidalgo et al. 2021). However, their potential role in such hydrocarbon-contaminated environments is still not clear.
A phylogenetic tree was reconstructed using the 20 MAGs with high and medium quality obtained in this study and 42 genomes from public databases (Table S5 and Fig. 1a). Taxonomic classification revealed that MAGs belonged to seventeen different families and twenty genera (Fig. 1a). MAGs affiliated at the species level were identified as Acinetobacter radioresistens MAG68 (Completeness 87% / Contamination 1.1%) and Moraxella cinereus MAG99 (Completeness 99.71% / Contamination 1.53%), both belonging to the family Moraxellaceae.
These results are consistent with the ones observed by the gene-centric analysis, where these families were shown to be the most abundant ones in the aquifer contaminated soils(Hidalgo et al. 2019)4. A. radioresistensis was found in high abundance in the monitoring well 62 (Fig. 1b, Family Moraxellaceae), which is located at the fringe of the plume (see Fig. 1a and Fig. 1 of Hidalgo et al., 2020). A. radioresistensis has been isolated from oily sludge and petroleum polluted soils, and its capability of degradation of a wide range of hydrocarbons and to produce bioemulsifier was reported in many studies(Mujumdar et al. 2019).
Seventeen MAGs were not identified at the species level, indicating that these correspond to putative novel lineages. These results demonstrate that hydrocarbon-polluted groundwater microbiomes are still poorly understood and that, in the case of this plume, the main players of aromatic hydrocarbon degradation are uncultured microbes. For example, three MAGs belonged to the Ignavibacteria class, from families Melioribacteraceae (n = 1), Ignavibacteraceae (n = 1) and SJA-28. Melioribacteraceae-related MAG was the most abundant microorganism in the present study (Fig. 1b). The Melioribacteraceae family is largely uncharacterized, with only one described and cultivated genus(Podosokorskaya et al. 2013). The phylum Ignavibacteria is closely related with Bacteoridetes/Chlorobi group (now Bacteroidota), which have been proposed as putative scavengers feeding on dead cells (Fig. 1b).
Three MAGs (MAG37, MAG58 and MAG95) were affiliated only at Bacteroidales order level, with relative abundances ranging from 1–8.5% in all BACTRAP (Fig. 1b). Previously studies demonstrated that uncultivated and novel lineages of Bacteroidetes potentially have syntrophic interactions with methanogenetic archaea in heavy oil contamination sites, producing acetate, propionate, formate and hydrogen, which can be used by methanogens for methane production(Xia et al. 2016; Dahle et al. 2008; Xiao-zhu. 2010). In addition, we were also able to identify a MAG assigned as Bacteroidales in a genus level (MAG83 - Flavobacterium genus), that was detected only in the PM62 metagenome (Fig. 1b). Several species from this genus have been reported as aerobic hydrocarbon degraders and as a dominant microbial community member in petroleum-contaminated sites(Agustini et al. 2020; Brown et al. 2015).
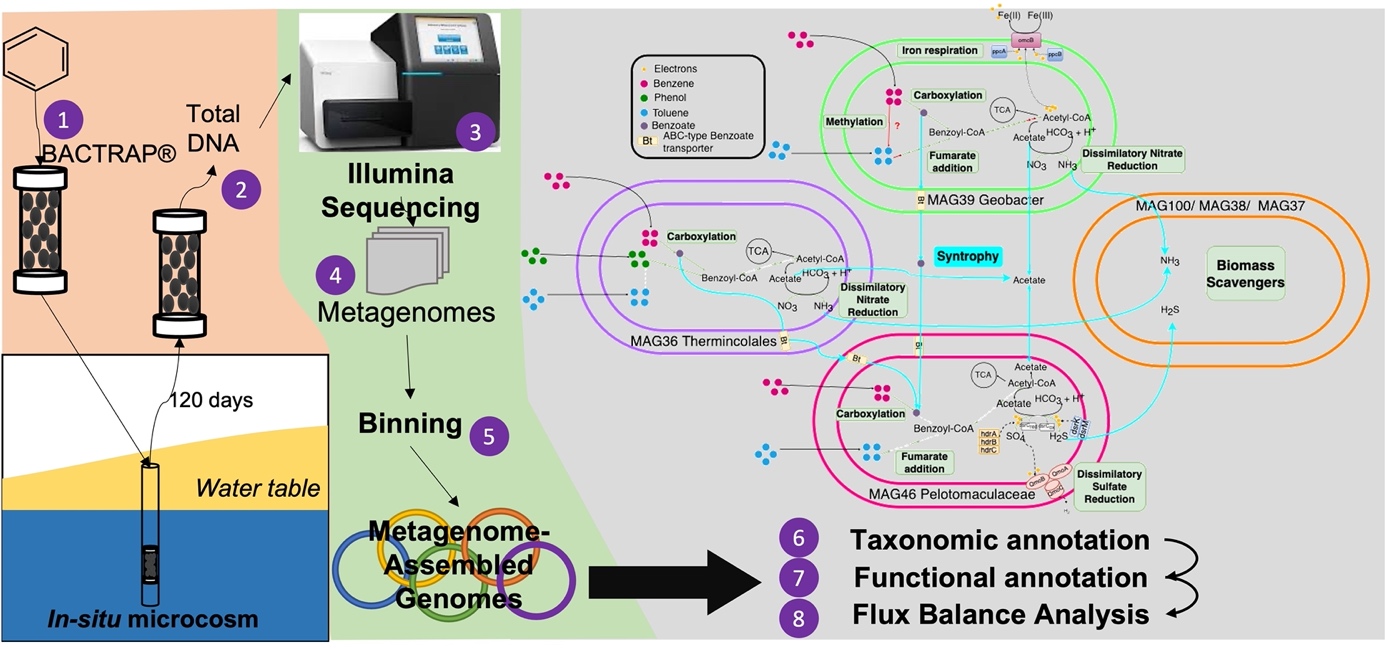
The only representative genome from the Desulfobacterota phylum was MAG39, assigned to Geobacter genus. It was found in high relative abundance in the BACTRAP® amended with benzene (Fig. 1b). This genus has been reported to activate benzene by hydroxylation, although it is widely known as being able to degrade aromatic hydrocarbons coupled to iron reduction by fumarate addition to toluene(Aburto-Medina and Ball. 2015; Abu Laban et al. 2010).
Finally, two MAGs from the phylum Firmicutes were obtained with high relative abundance in benzene-amended in-situ microcosms (Fig. 1b), named MAG36 and MAG46, affiliated to the order Thermincolales and family Pelotomaculaceae, respectively. According to the old classification, these two MAGs would belong to the order Clostridiales and to the family Peptococcaceae. Currently, based on the Genome Taxonomy Database (GTBD), Firmicutes includes the classes Desulfotomaculia and Thermincolia, which MAG46 and MAG36 were affiliated to, respectively. The phylogenetic tree showed that Thermincolales MAG36 is distantly related to Thermincola ferriatica e T. potens (Fig. 1a). However, it was not possible to determine which family and genus this MAGs belongs to. Several studies have demonstrated that different strains of Thermincola are able to degrade benzene under nitrate-reducing or iron-reducing conditions(Abu Laban et al. 2010; Toth et al. 2021). Results also showed that Pelotomaculaceae-affiliated MAG46 is distantly related to Pelotomaculum propionicicum. Some members of the genus Pelotomaculum are obligate syntrophs, that only can oxidize organic substrates as benzene in the presence of an electron-accepting partner(Kunapuli et al. 2007; Dong et al. 2017; Atashgahi et al. 2018; Steffi et al. 2010). On the other hand, with regards to the other MAGs belonging to Kapabacteriales order, Patesceibacteria phylum and Nitrosotenuis genus, their relationship with polluted environments is yet unknown.
Four MAGs from the recent proposed phylum Patesceibacteria were reconstructed from the in-situ microcosm metagenomes. This phylum is characterized by small cell and genome sizes(Tian et al. 2020; Hidalgo et al. 2021), carrying only essential genes and lacking a lot of metabolic functions(Hidalgo et al. 2021; Brown et al. 2015). For this reason, some authors have proposed that members of this phylum have a symbiont lifestyle(Castelle et al. 2018; Hidalgo et al. 2021).
3.2. Reconstruction of potential metabolisms
In total, 214 specific genes associated with hydrocarbon degradation traits were filtered in the functional annotation with KEGG database (Table S3). Additionally, CANT-HYD and PFAM databases were used in order to find the bacterial benzene carboxylase (AbcA)(An et al. 2013)d type polyheme cytochrome-C7 omcB domain, which is an important component in electron transport to Fe(III)(Leang et al. 2003), respectively. Sixteen of the twenty MAGs had at least one specific gene. The percentage of completeness of each specific metabolic pathway was calculated and plotted (Fig. 2). No naphthalene degradation genes were found. Specific functional annotations revealed four potentially hydrocarbon degrading bacteria: Acinetobacter radioresistens (MAG68), Geobacter (MAG39), Thermincolales (MAG36) and Pelotomaculaceae (MAG46). The first one is an aerobic degrader with genes in the four aerobic pathways studied (alkane degradation, catechol meta-cleavage, catechol ortho-cleavage and ring cleavage via Baeyer-Villiger oxidation). The other three genomes showed genes from at least two anaerobic pathways. Geobacter MAG39 showed functional potential for the complete degradation of aromatic hydrocarbons, with 14 of the 16 genes coding for toluene degradation and all the 10 genes coding for reduction of benzoyl-CoA, a key intermediate in anaerobic aromatic degradation (Fig. 3). The Thermincolaes MAG36 had two of the five genes from phenol anaerobic degradation pathway and six of the 10 genes from benzoyl-CoA degradation pathway, so it is possible that this microbe can degrade phenol but not toluene. The Pelotomaculaceae MAG46 showed a lower number of genes from each anaerobic pathway, one for benzoyl-CoA and two for phenol and toluene anaerobic degradation (Fig. 2). Therefore, this MAG is less complete than the other two degrading MAGs obtained (Table S2). Another interesting fact is the presence of 100% of the genes of dissimilatory sulfate reduction pathway in Pelotomaculaceae MAG46. Members of this family are known as fermenters, able to degrade hydrocarbon by syntrophy with anaerobic respiring microorganisms. However, in this study, Pelotomaculaceae MAG46 showed genetic potential for complete sulfate reduction(Dong et al. 2017).
According to the results of our previous study, phylotypes affiliated to the genera Geobacter and Pelotomaculum are key organisms in the biodegradation process occurring in the jet-fuel polluted plume(Hidalgo et al. 2019). In the present study, we obtained three MAGs with metabolic potential to degrade aromatic hydrocarbons anaerobically, MAG39 (genus Geobacter), MAG46 (family Pelotomaculaceae) and MAG36 (order Thermincolales) (Fig. 3).
3.2.1. Phenol degradation
Genes of phenol degradation (bsdB and hbaBCD) were found in the three potentially anaerobic degrading MAGs, suggesting capabilities for phenol degradation (Fig. 3). The Phenol degradation is mediated by the enzymes flavin prenyltransferase (bsdB), 4-hydroxybenzoate-CoA ligase (hbaA) and 4-hydroxybenzoyl-CoA reductase (hbaBCD)(Abu Laban et al. 2009). This potential to degrade phenol may suggest benzene degradation via hydroxylation as activation mechanism (Fig. 3). Although the enzyme for benzene hydroxylation to phenol is still unknown, Zhang and collaborators (2013) found evidence of benzene hydroxylation by G. metallireducens. They used H218O-labelled water and knocked out phenol degradation genes in G. metallireducens. The authors observed production of O18-labelled phenol, suggesting that the enzyme for hydroxylation can incorporate the hydroxyl group from water without molecular oxygen involved(Zhang et al. 2013).
3.2.2. Benzene degradation
Thermincolales MAG36, Geobacter MAG39 and Pelotomaculaceae MAG46 also have the functional potential to degrade benzene via carboxylation due to the presence of the putative anaerobic benzene carboxylase encoded by the abcA gene and the benzoate-CoA ligase (Fig. 3) for conversion to benzoate-CoA and benzoyl-CoA, respectively. Hitherto, there are no reports in the literature about the presence of gene abcA in Geobacter. On the other hand, the ability of Geobacter members to degrade benzoate has already been described in previous works. Simon et al. (2005) studied the gene cluster involved in benzoate degradation in Geobacter metallireducens(Simon et al. 2005). Peptococcaceae MAG46 showed potential to degrade only benzene and not benzoate, by the presence of the abcA gene. However, Thermincolales in MAG36 have both genes in its genome. Luo et al. (2014) studied benzene degradation by a nitrate-reducing enrichment culture using the metatranscriptomic approach. They showed that the first attack to the ring was performed by a Peptococcaceae member (now Pelotomaculaceae) producing benzoate, which was further transformed to benzoyl-CoA by a benzoate-degrading denitrifying Azoarcus strain, suggesting a syntrophic association between both bacteria(Luo et al. 2014; Atashgahi et al. 2018). In the same way, Kunapuli and collaborators (2007) suggested the primary oxidation of benzene by a Peptococcaceae strain and then further transformations by a syntrophic relationship with an iron-reducing Desulfobulbaceae(Kunapuli et al. 2007). Similar to these results, in the study performed by Toth and colleagues (2021), a syntrophic relationship between a Thermincola strain and nitrate-reducing bacteria to degrade benzene via carboxylation was observed(Toth et al. 2021).
3.2.3. Toluene degradation
The bss gene was not found in the Geobacter assembled-genome (Fig.s 2, Fig. 3). Nevertheless, most of the genes of the peripheral pathway to yield benzoyl-CoA were found in the genome (Fig. 3), suggesting a putative capacity to degrade toluene via the fumarate addition pathway. In MAG46, affiliated to family Pelotomaculaceae, bss and bbsH genes were found. Since this genome is 72.04% complete, we hypothesized that the other genes from the toluene degradation pathway via fumarate addition could not be recovered. However, this microorganism has the metabolic potential to perform this pathway, corroborating its role as primary degrader as observed in some studies(Luo et al. 2014; Abu Laban et al. 2010; Atashgahi et al. 2018). In the reconstruction of Pelotomaculum candidate BPL genome, the bss gene was not detected(Dong et al. 2017). Nevertheless, the capacity for toluene degradation by members of the Pelotomaculaceae family has been previously confirmed in oil polluted sediments by carbon incorporation from isotopically labelled-toluene into the biomass of Pelotomaculaceae species(Winderl et al. 2010). To our knowledge, MAG46 is the first reconstructed genome belonging to the family Pelotomaculaceae containing the bss gene. These findings indicate that microbial communities on the benzene- and toluene-amended BACTRAP®s have the metabolic potential to degrade toluene via fumarate addition. This corroborates the results obtained in our previous work, where a high abundance of bbsABC and bbsEF genes in these BACTRAP®s were found(Hidalgo et al. 2019), suggesting that toluene was degraded by microorganisms using the fumarate addition pathway.
3.2.4. Benzoyl-CoA degradation
Genes involved in the conversion of benzoyl-CoA (i.e., bamBC, bcrABC) were found in the three anaerobic degrading MAGs (MAG36, MAG39 and MAG46). Reduction of the key intermediate of anaerobic aromatic degradation, benzoyl-CoA, can be catalyzed by two distinct types of benzoyl-CoA reductases (BCRs), named class I ATP-dependent and class II ATP-independent BCR. The class I ATP-dependent BCR (encoded by the bcr genes) is present in facultative anaerobes (e.g., Thauera aromatica), and the class II ATP-independent BCR (encoded by the bam genes) is present exclusively in strictly anaerobic bacteria, such as Geobacter species(von Netzer et al. 2016; Fuchs et al. 2011; Löffler et al. 2011). Class I and II BCR genes were detected in the MAGs affiliated to Geobacter and to Thermincolales (Fig. 4) and only bam genes in the MAG affiliated to family Pelotomaculaceae. This is in accordance with the previously observed presence of class II BCR bam genes in G. metallireducens(Simon et al. 2005; Kung et al. 2009). Genes bamDEFGHI were identified at least in one copy in the genome of the Pelotomaculum candidate BPL genome(Dong et al. 2017; Simon et al. 2005).
Genes for the complete benzoyl-CoA β-oxidation-like pathway (i.e., dch, had, oah, fadABNJ, gcdACD, paaFH, atoB) were mainly found in the Geobacter MAG39. This finding is corroborated by the study of Kim et al (2013), where DNA stable isotope probing, and total metagenome sequencing were used to analyze the functional potential of iron-reducing bacteria involved in the toluene anaerobic degradation. The authors observed the presence of all genes for benzoyl-CoA conversion to CO2(Kim et al. 2014). Dong and co-workers reconstructed the genome of a Pelotomaculum candidate BPL and found a complete benzoyl-CoA β-oxidation-like pathway(Dong et al. 2017). However, in MAG46 (Pelotomaculaceae member) only some genes of the benzoyl-CoA degradation pathway were found, such as gene acd for dehydrogenation, as well as genes responsible for further β-oxidation until acetyl-CoA, i.e. 3-hydroxyacyl-CoA dehydrogenase (fadBJN), glutaryl-CoA dehydrogenase (non-decarboxylating) (acd), and acetyl-CoA C-acetyltransferase (atoB).
3.2.5. Sulfate, nitrate and iron reduction coupled to hydrocarbon degradation
Under anaerobic conditions, hydrocarbons are degraded coupled to respiration of electron acceptors after depletion of oxygen depending on the redox potential and energy yield (O2 > NO3− > Fe(III) > SO4- > CO2)(Ladino-Orjuela et al. 2016). For this reason, genes related to dissimilatory nitrate and sulfate reduction were searched in the MAGs. Genes encoding for nitrate (narGHI) and nitrite reductase (nrfAH) of the dissimilatory nitrate reduction pathway (nitrate to ammonia) were found in the Geobacter MAG39. G. metallireducens has the ability to use nitrate as electron acceptor besides respiring ferric iron(Senko and Stolz. 2001). Iron reduction ability is the main characteristic of members belonging to the Geobacter genus. The iron reduction system is responsible for the transport of electrons released by the oxidation of organic compounds in the cytosol through the periplasmic space outside the cell. These electrons are finally transferred to ferric iron (Fe(III)), that is reduced to ferrous iron (Fe(II)) (Fig. 4). The complete electron transport chain was studied in Geobacter sulfurreducens and is composed of c-type multiheme cytochromes encoded by 110 genes(Leang et al. 2003). The transport of electrons is based on the redox potential of each cytochrome. The metal-reduction-associated cytochrome (MacA) transfers electrons from the cytosol to the periplasmic c-type cytochrome (PpcA). The outer membrane cytochromes (OMCs) transport the electrons to the extracellular acceptor. Different OMCs have been described depending on the type of electron acceptor. OmcB is a c-type polyheme cytochrome essential for Fe(III) reduction(Leang et al. 2003; Kracke et al. 2015). MacA, PpcA and OmcB were found in Geobacter MAG39, confirming its potential to use Fe(III) as electron acceptor and to produce Fe(II) (Fig. 4). This result agrees with the high relative abundance of the omcB gene in the sequenced metagenomes of the microbial communities colonizing the in situ microcosms and with the physicochemical characterization of the plume and source zone, that revealed iron (Fe2+) as the most abundant ion, suggesting that iron-reducing conditions were prevalent in the polluted aquifer(Hidalgo et al. 2019).
Potential for sulfate reduction was observed only in Pelotomaculaceae-MAG46. The pathway starts with sulfate activation by sulfate adenylyl transferase (sat), producing adenosine 5’phosphosulfate (APS)(Taguchi et al. 2004), which is reduced to sulfite by adenylsulfate or APS reductase encoded by aprAB genes (Fig. 4)(Fritz et al. 2002). Finally, sulfite is reduced to sulfide by dissimilatory sulfite reductase (dsrAB) (Fig. 4)(Dahl et al. 1993), with the involvement of the small protein DsrC(Cort et al. 2001; Oliveira et al. 2008; Pires et al. 2006). DsrD is another small protein, which is encoded downstream of dsrAB genes, possibly also involved in the sulfite reduction with a regulatory role(Mizuno et al. 2003). Imachi et al. (2006) suggest that Pelotomaculum members are not able to reduce sulfate(Imachi et al. 2006). Nevertheless, well known sulfate reducing members of the genus Desulfotomaculum are a close relative. Additionally, a sulfate reducing benzene-degrading Pelotomaculum phylotype has been described(Abu Laban et al. 2009), and sulfate reducinge genes were also found in the genome reconstruction of the candidate Pelotomaculum BPL(Dong. 2017).
QmoABC and dsrMKJOP are gene clusters coding for two membrane complexes proposed as electron donors of the reductases AprAB and DsrAB, respectively(Pereira et al. 2008). Desulfovibrio desulfuricans was the first bacterium described to have the QmoABC (quinone-interacting membrane-bound oxidoreductase complex) system(Pires et al. 2003), that includes three subunits binding two hemes b, two FAD groups and several iron-sulfur centers. QmoA and QmoB are homologous to HdrA, a flavin-containing subunit of the soluble heterodisulfide reductases(Hedderich et al. 2005). QmoC is a fusion protein that contains a cytochrome b transmembrane domain related to HdrE (membrane-bound cytochrome b) and a hydrophilic iron-sulfur domain related to HdrC (small iron-sulfur protein)(Pereira et al. 2011). It has been proposed that the electrons are transferred from the Qmo pool to AprAB, since the qmo genes were found adjacent to aprAB (Fig. 4). This process may result in energy conservation(Pires et al. 2003; Venceslau et al. 2010). Pereira and collaborators (2011) performed a comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea, confirming that the qmoABC genes are present in the majority of the sulfate reducing organisms analyzed(Pereira et al. 2011). The Gram positive bacteria Desulfotomaculum acetoxidans and Candidatus Desulforudis audaxviator are exceptions where the qmoC gene is absent and replaced by hdrBC genes that code for soluble subunits of HDRs(Junier et al. 2010), suggesting that in Gram-positive bacteria electrons for APR reductase may come from other mechanism rather than quinones, without energy conservation(Pereira et al. 2011). Genes coding for the qmo complex were detected in MAG46, as well as hdr genes (Fig. 4). On the other hand, the DsrMKJOP system is a transmembrane complex with redox subunits. Thus, DsrJ (triheme cytochrome c) and DsrO (FeS protein) are localized in the periplasm, whereas DsrM (diheme cytochrome b) and DsrP (quinone-interacting protein) are in the membrane and, finally, DsrK (iron-sulfur protein) is in the cytoplasm(Pires et al. 2006; Pereira et al. 2011). In Gram positive bacteria, only dsrMK is present (Fig. 4), indicating that only the two corresponding proteins are essential for sulfite reduction. DsrMK can oxidize menaquinol and reduce DsrC disulfide, donating electrons from a DsrAB enzyme to sulfite reduction(Oliveira et al. 2008). Both dsrMK and dsrC genes were found in Pelotomaculaceae-MAG46. Based on the presence of aprAB, dsrAB, qmoABC, drsMK and dsrC genes, we propose that MAG46 has potential for the complete sulfate reduction pathway during anaerobic degradation of benzene and toluene. However, Pelotomaculaceae members were also proposed as being able to degrade benzene under sulfate reduction conditions in a hypothesized syntrophic process, where this bacterium degrade benzene by fermentation coupled to sulfate reduction performed by another microorganism(Rakoczy et al. 2011) 70, 100. Abu Laban and collaborators reported the first Pelotomaculum phylotype able to degrade benzene coupled to sulfate reduction without a syntrophic partner organism(Abu Laban et al. 2009). The genome here reconstructed supports the hypothesis that the corresponding organism is a sulfate reducing benzene-degrading bacterium, due to the presence of the complete sulfate reduction pathway (Fig. 4).
3.2.6. Aerobic aromatic hydrocarbon degradation
The aerobic aromatic hydrocarbon degradation has three stages: i) initial attack or activation, ii) de-aromatization or ring cleavage, and iii) further degradation by lower pathways. The activation can be performed by mono- or di-oxygenases enzymes. At this point, most of aerobic degraders produce the central intermediary catechol. Then, this compound can be cleaved either ortho or meta cleavage by intra or extradiol dioxygenases. Finally, the intermediates from de-aromatization of catechol are oxidized until central metabolites as pyruvate, acetyl-CoA, or succinyl-CoA(Jindrová et al. 2002).
In this study, only one MAG (MAG68) with ability to degrade aromatic hydrocarbons aerobically was obtained, containing Acinetobacter radioresistens (family Moraxellaceae). This MAG was found in high abundance in the monitoring well PM62, located at the fringe of the plume(Hidalgo et al. 2019), and showed metabolic potential to degrade catechol via ortho or meta cleavage (catA) and alkanes (alkB). Caposio et al. (2002) studied two catechol 1,2-dioxygenase genes from a strain of Acinetobacter radioresistens, able to grow on phenol and benzoate as sole carbon and energy source(Caposio et al. 2002). Acinetobacter has been widely studied in wastewater treatment due to its ability for degradation of hydrocarbons, mainly phenol and short chain alkanes (C9-C14)(Wu et al. 2017).
The current results are consistent with the previous diversity analysis of the relative abundance based on 16S rRNA amplicon sequencing, where Moraxellaceae was the most abundant family at the fringe of the plume(Hidalgo et al. 2019). Additionally, the gene-centric approach showed that some hydrocarbon degradation genes, such as dmpN, dmpO, catA and alkB2, were assigned to this family, suggesting benzene degradation via phenol to catechol, and alkane degradation(Hidalgo et al. 2019). Finding aerobic microorganisms and metabolisms was an unexpected result, due to the presence of a LNAPL source zone that is characterized to be an anoxic environment. However, it was hypothesized that the presence of aerobic microorganisms is a result of vertical movement of the water table over an annual cycle, creating an extensive smear zone at the water table with dynamically changing biogeochemical conditions. Oxygen entrapment in sediment when the water table rises is suggested to by responsible for temporal aquifer oxygenation(Hidalgo et al. 2019; Teramoto and Chang. 2019; Haberer et al. 2015).
3.3. Flux Balance Analysis
In order to explore the potential interactions and metabolic exchange between the MAGs in benzene- and toluene-amended BACTRAP®s, a flux balance analysis (FBA) was performed. To calculate the metabolic models, MAGs with high abundance in each sample were used (Table S6) and the media composition for gap filling was M9[-O2] (anaerobic minimal medium), with minor changes (Table S4, Supplementary information 1). FBA results (Fig. S3) showed a main cross-feeding interaction based on nitrite, with benzoate and H2S as e− donors. Additionally, the interactions with a Smetana score > 0.2 in these two samples were plotted as networks (Fig. S4) in order to build the cross-feeding interaction model, where is shown which microorganisms are the donors and/or recipients of these metabolites.
Nitrite, which is an intermediate of the nitrate dissimilatory pathway used by some microorganisms coupled to the degradation of hydrocarbons, was mainly exchanged between Geobacter MAG39 with Melioribacteriaceae MAG100, Kapabacteriales MAG38 and Bacteroidales MAG37. A genome-scale constraint model of G. metallireducens built by Sun et al. (2009) showed that this microorganism can use nitrate as an electron acceptor(Sun et al. 2009). They simulated the growth of G. metallireducens with nitrate as electron acceptor and acetate, ethanol, pyruvate and benzoate as electron donor, and observed that the highest biomass yield was obtained with nitrate and benzoate(Sun et al. 2009).
Benzoate was also important in the cross-feeding interactions, especially in the benzene-amended BACTRAP (B65). Benzoate is the intermediate of benzene activation via carboxylation. Pelotomaculaceae MAG46 was the main recipient of benzoate. The ABC-type benzoate transporter gene was found in the genome sequence of MAG46 (Fig. 4). Konishi Nobu and collaborators (2017) used metagenomics and metatranscriptomics to characterize the in situ catabolic behavior and diversity of syntrophic metabolizers (Syntrophus, Pelotomaculum and Syntrophorhabdus) during the degradation of three different aromatics, including benzoate. They observed that Pelotomaculum was able to degrade benzoate(Nobu et al. 2017), despite the lack of the ABC-type benzoate transporter gene (K01999) in the genome. However, the important role of the ABC-type transporter in the degradation of different aromatic hydrocarbons (e.g., benzoate and 4-hydroxybenzoate) was confirmed in other species such as Pseudomonas huttiensis and Rhodopseudomonas palustris (Giuliani et al. 2011; Yuroff et al. 2003).
Another main compound, especially in the cross-feeding interactions of the toluene model, was hydrogen sulfide (H2S), that is the product of sulfate dissimilatory reduction, when sulfate is used as electron acceptor for hydrocarbon degradation. As it was already discussed above, Thermincolales MAG36 (Family UBA2595, before Peptococcaceae) has metabolic potential to reduce sulfate to H2S. Based on the model, sulfide was exchanged mainly with Melioribacteriaceae MAG100 and Patescibacteria MAG59. Abu Laban et al. (2009) and Dong et al. (2017) proposed that some members of the family Peptococcaceae are sulfate reducers and are not obligate syntrophs, as some species of this family(Dong et al. 2017; Abu Laban et al. 2009; Dong. 2017). These results are corroborated by our findings based on the genomic analysis of MAGs about the metabolic potential of the microbial community to degrade benzene via carboxylation and toluene coupled to nitrate and sulfate reduction.
As discussed above, MAG100, MAG38 and MAG37 were the main recipients of final anaerobic respiration products, such as nitrite and H2S. Members of class Ignavibacteria and phylum Bacteroidota have been proposed as secondary degraders that scavenge dead biomass or hydrocarbon intermediates(Gieg et al. 2014).
{kind=link}